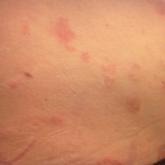
Article Autoimmune Progesterone Dermatitis Author: Ivy DeRosa, DO Brett Bender, DO Michael Centilli, DO Read More
Article Erythematous patches on the hands Author: David A. Kasper, DO, MBA Michael Centilli, DO Kimball Silverton, DO Our patient’s history of obsessive-compulsive disorder may have masked the true diagnosis for years. Read More