User login
Fever, tachycardia, and tachypnea during a psychotic exacerbation
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.

Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.

EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
Treated with a mood stabilizer, he becomes incontinent and walks oddly
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
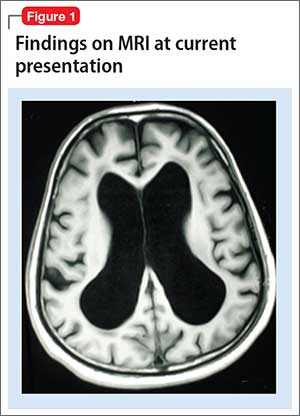
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
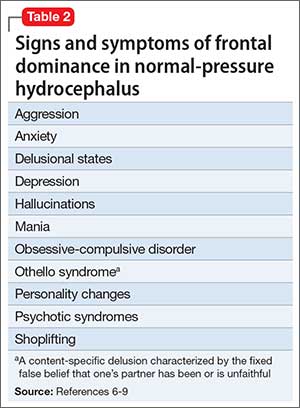
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.

Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
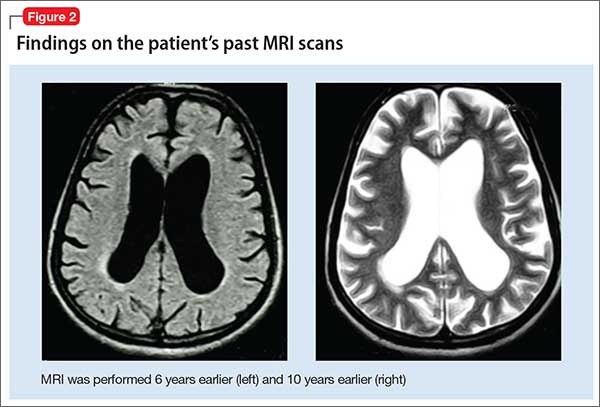
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
Depressed and confused, and dizzy while walking the dog
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
Dissecting melancholia with evidence-based biomarker tools
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).

Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).


In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
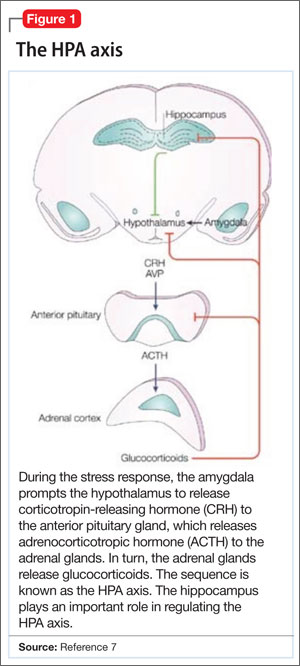
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
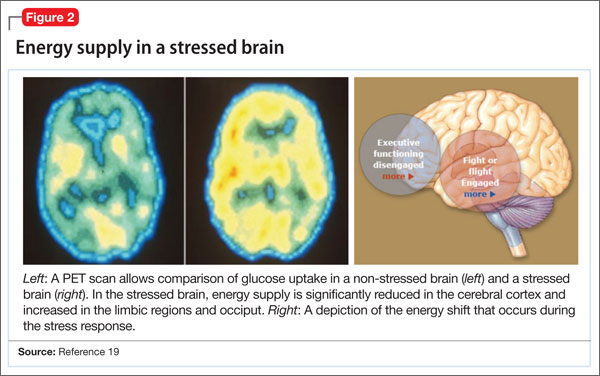
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
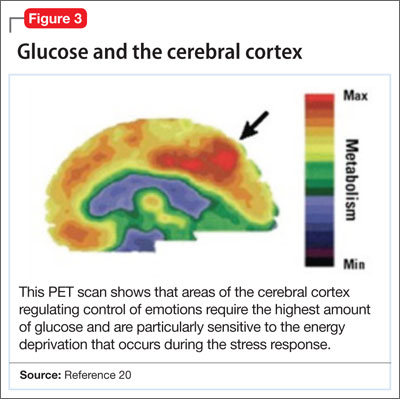
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
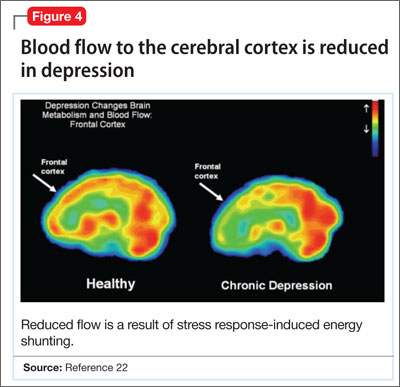
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).

Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
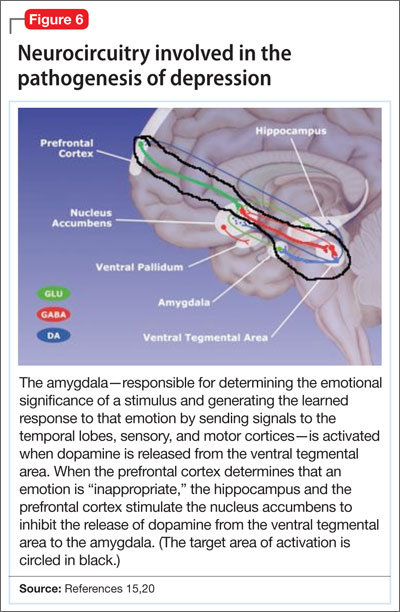
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
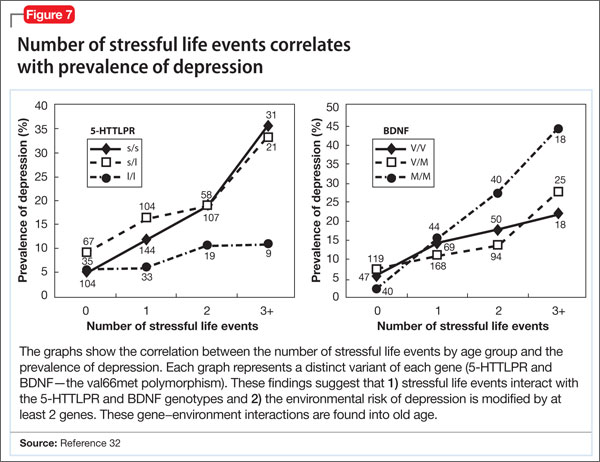
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.

Conversion disorder? One patient’s ‘moving’ story
History: 3 ‘Uncontrollable’ months
Ms. M, age 57, presents to the ER complaining of coordination problems and involuntary limb movements that have gradually worsened over 3 months.
Two months ago, Ms. M’s primary care physician and neurologist diagnosed her with conversion disorder. Brain MRI at the time showed mild chronic ischemic changes; cervical spinal cord MRI was normal. The neurologist referred Ms. M to a psychiatrist, who prescribed duloxetine, dosage unknown. She started having suicidal thoughts and trembling after starting the medication, so she stopped taking it after 1 week.
Physical exam shows upbeat nystagmus, inconsistent sensory findings, limb ataxia that is more pronounced on the right side, and uncontrollable limb movements, particularly of the right arm.
Ms. M is divorced, lives alone, and works as a medical secretary. Four months ago, she marked the fifth anniversary of her daughter’s death from a drug overdose at age 20. Her parents, whom she cared for, died within the last 3 years. Her son recently left home to attend graduate school, and she is estranged from the rest of her family. She endorses depressed mood and grief over her daughter’s death but says she has no one with whom to talk. She also feels persistent guilt, as she was out on a date when her daughter tried to call home shortly before her death.
The limb movements and lack of coordination are increasingly interfering with Ms. M’s life. She often uses her left hand to stop the right from moving and to guide it in simple tasks, such as opening doors. She can no longer hold a cup of coffee in her right hand or stand on stools at work to reach overhead shelves. At presentation, Ms. M’s imbalance and involuntary movements are so severe that she cannot walk. A coworker drove her to the ER.
poll here
The authors’ observations
A neurologist who evaluates Ms. M in the ER is concerned about her vertical nystagmus, which, unlike horizontal nystagmus, is almost always pathologic. The neurology service admits her for further evaluation.
Ms. M’s age, recent normal MRIs, physical presentation, and lack of other findings suggest a paraneoplastic syndrome. Ataxia associated with subacute cerebellar degeneration can indicate an occult malignancy and is closely linked to gynecologic and breast cancers. Cerebellar degeneration often begins with loss of coordination, can be unilateral, and can appear as intention myoclonus.1
Also considered are:
- opsoclonus-myoclonus, which presents with ataxia, myoclonus, and random chaotic eye movements. This paraneoplastic disorder is less common in adults than in children, however.1
- alien hand/limb syndrome, in which the limb unintentionally performs seemingly purposeful movements, often prompting the patient to restrain the limb with the other hand. This syndrome, however, usually localizes to a lesion in the medial frontal lobe or corpus callosum. Ms. M’s brain MRIs show no such lesion.
poll here
Treatment: Searching for answers
We order an extensive neurologic workup for Ms. M, focusing on causes of inherited and acquired ataxias. The evaluation includes:
- brain and cervical spine MRIs to check for focal cerebral and spinal lesions
- EEG to search for seizure activity and slowing characteristic of encephalopathies
- urine heavy metal testing for toxic processes
- thyroid-stimulating hormone testing for hypothyroid-associated ataxia.
Paraneoplastic workup includes chest, pelvic, and abdominal CT; a gynecologic exam; and a mammogram. All results are negative or equivocal.
We also order blood tests for paraneoplastic antibodies, evidence of opsoclonus/myoclonus, and spinocerebellar ataxia genetic testing; and a CSF check for protein 14-3-3 levels suggestive of prion disease. These tests, run at specialized laboratories, take 4 to 6 weeks.
Ms. M remains hospitalized for 7 days for evaluation. Her movement problems persist, though they often abate when she is distracted. Her upbeat nystagmus appears intermittent. Her affect is diverse, often shifting between tearfulness and inappropriate laughter.
Based on interviews with Ms. M, the C/L team sees prominent depressive symptoms including marked difficulty sleeping, appetite loss, and excessive guilt over her daughter’s death. She also seems indifferent towards her disabling motor symptoms.
The C/L team diagnoses Ms. M with chronic and acute adjustment disorder and major depressive disorder. She is initially hesitant to take another antidepressant but agrees to try mirtazapine, 15 mg nightly, to treat her depression, decreased appetite, and sleep problems. After 2 days, mirtazapine is increased to 30 mg nightly as she is tolerating it and is willing to try a higher dosage.
poll here
The authors’ observations
No neurologic or pathologic explanation is found for Ms. M’s symptoms. Imaging reveals no lesions to explain her intermittent upbeat nystagmus, which localizes to the pons and caudal medulla.2
Conversion disorder. Ms. M, however, appears to meet DSM-IV-TR criteria for conversion disorder (Box), which is thought to result from intense psychological distress in persons who can only express such emotions somatically. Her complaints had specific precursors: she was newly separated from her son and had marked the anniversary of her daughter’s death, which intensified her persistent mourning. We link both circumstances temporally to symptom onset. Also, lack of interest in her serious motor symptoms could be the “la belle indifference” typical of conversion disorder.
Ms. M, however, appears highly suggestible. Her physical symptoms improve soon after her attending psychiatrist suggests that treating her depression will decrease her movements. The neurologists also notice day-to-day fluctuations in her gait disturbance and jerking movements. Distraction techniques produce objective improvement in both symptoms.
- One or more symptoms or deficits affecting voluntary motor or sensory function that suggest a neurological or other general medical condition.
- Psychological factors are judged to be associated with the symptom or deficit because the initiation or exacerbation of the symptom or deficit is preceded by conflicts or other stressors.
- The symptoms or deficit is not intentionally produced or feigned (as in factitious disorder or malingering).
- The symptom or deficit cannot, after appropriate investigation, be fully explained by a general medical condition, or by the direct effects of a substance, or as a culturally sanctioned behavior or experience.
- The symptom or deficit causes clinically significant distress or impairment in social, occupational, or other important areas of functioning or warrants medical evaluation.
- The symptom or deficit is not limited to pain or sexual dysfunction, does not occur exclusively during the course of somatization disorder, and is not better accounted for by another mental disorder.
Specify type of symptom or deficit:
With motor symptom or deficit
With sensory symptom or deficit
With seizures or convulsions
With mixed presentation
Source: Diagnostic and statistical manual of mental disorders (4th ed-text rev). Copyright 2000.
American Psychiatric Association. Reprinted with permission.
In malingering, the patient seeks external incentives for feigned behavior. The role of secondary gain must be considered, as Ms. M’s illness has reunited her with her son, who visits her regularly at the hospital.
Ms. M’s evaluation, however, uncovers no evidence that she is intentionally producing symptoms.
Follow-up: The answer becomes clear
One month after discharge to inpatient rehabilitation, Ms. M is readmitted to the neurology unit. Her uncontrollable limb jerks and ataxia are worse, and she appears demented and near mute. At that time, we learn that the CSF sample sent during her first admission is positive for protein 14-3-3.
Ms. M is diagnosed with Creutzfeldt-Jakob disease (CJD), a spongiform encephalopathy secondary to prion disease. She dies 6 days later. Sporadic CJD is confirmed at autopsy.
The authors’ observations
The literature lists no comprehensive differential diagnosis for conversion disorder, probably because presentations are diverse and the symptoms overlap with innumerable neurologic and medical conditions. This is underscored by the broad differential diagnosis for Ms. M’s ataxia.
In a study to identify organic syndromes initially diagnosed as conversion disorder,4 10 of 85 patients (11.8%) were initially misdiagnosed and later found to have dyskinesia, amyotrophic lateral sclerosis, multiple system atrophy, extrapyramidal syndrome, multiple sclerosis, dementia, Parkinson’s disease with psychogenic aggravation, lung cancer with cerebral metastases, and radicular syndrome. CJD and conversion disorder also share many symptoms (Table).
Correct diagnosis of conversion disorder calls for ruling out neurologic and medical conditions. Ms. M’s upbeat nystagmus prompted aggressive neurologic evaluation. Although horizontal nystagmus has been reported rarely in conversion disorder,5 vertical nystagmus has not. One case report6 describes vertical nystagmus as the first clinical sign of CJD.
Leading clinical symptoms of CJD include progressive dementia, myoclonus, cerebellar ataxia, visual problems, and extrapyramidal signs.7 Ms. M’s uncontrollable movements and jerks, although not classically myoclonic, were similar to this common finding. She did not present with dementia, but her rapidly progressive end-stage mental status changes were characteristic of CJD.
Sporadic CJD accounts for 84% of transmissible spongiform encephalopathies. Genetic, iatrogenic, and variant CJD forms (linked to bovine spongiform encephalopathy, or “mad-cow disease”) account for other cases.8 Psychiatric symptoms are a more-common manifestation of variant CJD9 but have been reported in sporadic CJD.10
Eventually, Ms. M’s upbeat nystagmus, persistent abnormal movements, rapidly progressive dementia, and elevated CSF protein 14-3-3 made the CJD diagnosis. Protein 14-3-3 is 94% sensitive and 84% specific for diagnosing CJD.11 Ms. M’s EEG findings did not suggest CJD, but these findings are less sensitive and occur later than the CSF findings.11
Finally, conversion disorder is almost always acute, not slowly progressive as with Ms. M.
Table
Conversion disorder, sporadic Creutzfeldt-Jakob disease share many symptoms
| Complaint | Conversion disorder | Sporadic CJD |
|---|---|---|
| Paralysis | May not follow motor pathways | No |
| Myoclonus | Yes | Cardinal manifestation |
| Ataxia | May be bizarre in character | Present in 25% to 30% of patients, reflecting multiple disease subtypes |
| Hyperreflexia | No | Yes (40% to 80% of patients) |
| Dysphagia | Yes | No |
| Vomiting | Yes | No |
| Aphonia | Yes | No |
| Diplopia | Yes | Rare |
| Nystagmus | Rare | Yes |
| Blindness | Hysterical blindness detectable by ophthalmologic examination | Rare |
| Deafness | Yes | Rare |
| Anesthesia | Yes | No |
| Paresthesia | Yes | No |
| Depression | Yes | Yes |
| Other psychiatry diagnoses | Yes | More common in variant CJD |
| Progressive dementia | No | Cardinal manifestation |
| Temporal relationship with stress | Yes | No |
| Left-side symptoms more common | Yes | No |
Getting the diagnosis right
DSM-IV-TR criteria state that conversion disorder symptoms cannot be otherwise explained “after appropriate investigation,” but what constitutes “appropriate” is unclear. Extensive inpatient evaluation eventually produced the correct diagnosis for Ms. M, but such a detailed evaluation may be too expensive and expansive for every patient with conversion disorder symptoms.
In the conversion disorder study,4 the 10 misdiagnosed patients received one to eight supplemental diagnostic techniques before being correctly diagnosed. In five of the patients, however, the general neurologic examination was identified as the diagnostic technique responsible for final diagnosis.
These findings suggest that a neurologic examination is key to evaluating complaints that suggest conversion disorder and to identify neurologic conditions. The results can also suggest somatic disorders, as exam findings will reflect patients’ perceptions of neurologic processes. For example:
- patients with conversion motor symptoms may have tonic contractures of antagonistic muscles to “paralyze” certain joints
- those with conversion sensory symptoms rarely have sensory impairments that follow known innervation patterns.
Motor complaints, such as localized paralysis or abnormal movements, should be evaluated with a brain MRI to look for lesions along the motor or cerebellar tracts. Sensory pathways can be further investigated with brain MRI and the relevant evoked potential(s) (visual, brainstem, or somatosensory).
Order EEG for patients with convulsions, particularly prolonged EEG monitoring with a video component, and measure serum prolactin immediately after an episode. In some cases, the neurologic exam alone or in conjunction with these initial studies can make the diagnosis. If the clinical situation warrants, more-detailed evaluations may be necessary.
Related resources
- Wise MG, Rundell JR. Clinical manual of psychosomatic medicine: a guide to consultation-liaison psychiatry. Arlington, VA: American Psychiatric Publishing; 2005.
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Duloxetine • Cymbalta
- Mirtazapine • Remeron
The authors report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Dropcho EJ, Dalmau J, Greenlee JE, et al. Paraneoplastic disorders: Central nervous system disorders. Continuum 1999;5:25-40.
2. Zingler VC, Strupp M, Jahn K, et al. Upbeat nystagmus as the initial clinical sign of Creutzfeldt-Jakob disease. Ann Neurol 2005;57:607-8.
3. Phillips KA (ed). Somatoform and factitious disorders. Washington, DC: American Psychiatric Publishing; 2001.
4. Moene FC, Landberg EH, Hoogduin KA, et al. Organic syndromes diagnosed as conversion disorder: identification and frequency in a study of 85 patients. J Psychosom Res 2000;49:7-12.
5. Smith CH, Beck RW, Mills RP. Functional disease in neuroophthalmology. Neurol Clin 1983;1:955-71.
6. Pierrot-Deseilligny C, Milea D. Vertical nystagmus: clinical facts and hypotheses. Brain 2005;128(Pt 6):1237-46.
7. Glatzel M, Stoeck K, Seeger H, et al. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005;62:545-52.
8. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005;64:1586-91.
9. Spencer MD, Knight RSG, Will RG. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ 2002;324:1479-82.
10. Jiang TT, Moses H, Gordon H, Obah E. Sporadic Creuztfeldt-Jakob disease presenting as major depression. South Med J 1999;92:807-8.
11. Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:811-15.
History: 3 ‘Uncontrollable’ months
Ms. M, age 57, presents to the ER complaining of coordination problems and involuntary limb movements that have gradually worsened over 3 months.
Two months ago, Ms. M’s primary care physician and neurologist diagnosed her with conversion disorder. Brain MRI at the time showed mild chronic ischemic changes; cervical spinal cord MRI was normal. The neurologist referred Ms. M to a psychiatrist, who prescribed duloxetine, dosage unknown. She started having suicidal thoughts and trembling after starting the medication, so she stopped taking it after 1 week.
Physical exam shows upbeat nystagmus, inconsistent sensory findings, limb ataxia that is more pronounced on the right side, and uncontrollable limb movements, particularly of the right arm.
Ms. M is divorced, lives alone, and works as a medical secretary. Four months ago, she marked the fifth anniversary of her daughter’s death from a drug overdose at age 20. Her parents, whom she cared for, died within the last 3 years. Her son recently left home to attend graduate school, and she is estranged from the rest of her family. She endorses depressed mood and grief over her daughter’s death but says she has no one with whom to talk. She also feels persistent guilt, as she was out on a date when her daughter tried to call home shortly before her death.
The limb movements and lack of coordination are increasingly interfering with Ms. M’s life. She often uses her left hand to stop the right from moving and to guide it in simple tasks, such as opening doors. She can no longer hold a cup of coffee in her right hand or stand on stools at work to reach overhead shelves. At presentation, Ms. M’s imbalance and involuntary movements are so severe that she cannot walk. A coworker drove her to the ER.
poll here
The authors’ observations
A neurologist who evaluates Ms. M in the ER is concerned about her vertical nystagmus, which, unlike horizontal nystagmus, is almost always pathologic. The neurology service admits her for further evaluation.
Ms. M’s age, recent normal MRIs, physical presentation, and lack of other findings suggest a paraneoplastic syndrome. Ataxia associated with subacute cerebellar degeneration can indicate an occult malignancy and is closely linked to gynecologic and breast cancers. Cerebellar degeneration often begins with loss of coordination, can be unilateral, and can appear as intention myoclonus.1
Also considered are:
- opsoclonus-myoclonus, which presents with ataxia, myoclonus, and random chaotic eye movements. This paraneoplastic disorder is less common in adults than in children, however.1
- alien hand/limb syndrome, in which the limb unintentionally performs seemingly purposeful movements, often prompting the patient to restrain the limb with the other hand. This syndrome, however, usually localizes to a lesion in the medial frontal lobe or corpus callosum. Ms. M’s brain MRIs show no such lesion.
poll here
Treatment: Searching for answers
We order an extensive neurologic workup for Ms. M, focusing on causes of inherited and acquired ataxias. The evaluation includes:
- brain and cervical spine MRIs to check for focal cerebral and spinal lesions
- EEG to search for seizure activity and slowing characteristic of encephalopathies
- urine heavy metal testing for toxic processes
- thyroid-stimulating hormone testing for hypothyroid-associated ataxia.
Paraneoplastic workup includes chest, pelvic, and abdominal CT; a gynecologic exam; and a mammogram. All results are negative or equivocal.
We also order blood tests for paraneoplastic antibodies, evidence of opsoclonus/myoclonus, and spinocerebellar ataxia genetic testing; and a CSF check for protein 14-3-3 levels suggestive of prion disease. These tests, run at specialized laboratories, take 4 to 6 weeks.
Ms. M remains hospitalized for 7 days for evaluation. Her movement problems persist, though they often abate when she is distracted. Her upbeat nystagmus appears intermittent. Her affect is diverse, often shifting between tearfulness and inappropriate laughter.
Based on interviews with Ms. M, the C/L team sees prominent depressive symptoms including marked difficulty sleeping, appetite loss, and excessive guilt over her daughter’s death. She also seems indifferent towards her disabling motor symptoms.
The C/L team diagnoses Ms. M with chronic and acute adjustment disorder and major depressive disorder. She is initially hesitant to take another antidepressant but agrees to try mirtazapine, 15 mg nightly, to treat her depression, decreased appetite, and sleep problems. After 2 days, mirtazapine is increased to 30 mg nightly as she is tolerating it and is willing to try a higher dosage.
poll here
The authors’ observations
No neurologic or pathologic explanation is found for Ms. M’s symptoms. Imaging reveals no lesions to explain her intermittent upbeat nystagmus, which localizes to the pons and caudal medulla.2
Conversion disorder. Ms. M, however, appears to meet DSM-IV-TR criteria for conversion disorder (Box), which is thought to result from intense psychological distress in persons who can only express such emotions somatically. Her complaints had specific precursors: she was newly separated from her son and had marked the anniversary of her daughter’s death, which intensified her persistent mourning. We link both circumstances temporally to symptom onset. Also, lack of interest in her serious motor symptoms could be the “la belle indifference” typical of conversion disorder.
Ms. M, however, appears highly suggestible. Her physical symptoms improve soon after her attending psychiatrist suggests that treating her depression will decrease her movements. The neurologists also notice day-to-day fluctuations in her gait disturbance and jerking movements. Distraction techniques produce objective improvement in both symptoms.
- One or more symptoms or deficits affecting voluntary motor or sensory function that suggest a neurological or other general medical condition.
- Psychological factors are judged to be associated with the symptom or deficit because the initiation or exacerbation of the symptom or deficit is preceded by conflicts or other stressors.
- The symptoms or deficit is not intentionally produced or feigned (as in factitious disorder or malingering).
- The symptom or deficit cannot, after appropriate investigation, be fully explained by a general medical condition, or by the direct effects of a substance, or as a culturally sanctioned behavior or experience.
- The symptom or deficit causes clinically significant distress or impairment in social, occupational, or other important areas of functioning or warrants medical evaluation.
- The symptom or deficit is not limited to pain or sexual dysfunction, does not occur exclusively during the course of somatization disorder, and is not better accounted for by another mental disorder.
Specify type of symptom or deficit:
With motor symptom or deficit
With sensory symptom or deficit
With seizures or convulsions
With mixed presentation
Source: Diagnostic and statistical manual of mental disorders (4th ed-text rev). Copyright 2000.
American Psychiatric Association. Reprinted with permission.
In malingering, the patient seeks external incentives for feigned behavior. The role of secondary gain must be considered, as Ms. M’s illness has reunited her with her son, who visits her regularly at the hospital.
Ms. M’s evaluation, however, uncovers no evidence that she is intentionally producing symptoms.
Follow-up: The answer becomes clear
One month after discharge to inpatient rehabilitation, Ms. M is readmitted to the neurology unit. Her uncontrollable limb jerks and ataxia are worse, and she appears demented and near mute. At that time, we learn that the CSF sample sent during her first admission is positive for protein 14-3-3.
Ms. M is diagnosed with Creutzfeldt-Jakob disease (CJD), a spongiform encephalopathy secondary to prion disease. She dies 6 days later. Sporadic CJD is confirmed at autopsy.
The authors’ observations
The literature lists no comprehensive differential diagnosis for conversion disorder, probably because presentations are diverse and the symptoms overlap with innumerable neurologic and medical conditions. This is underscored by the broad differential diagnosis for Ms. M’s ataxia.
In a study to identify organic syndromes initially diagnosed as conversion disorder,4 10 of 85 patients (11.8%) were initially misdiagnosed and later found to have dyskinesia, amyotrophic lateral sclerosis, multiple system atrophy, extrapyramidal syndrome, multiple sclerosis, dementia, Parkinson’s disease with psychogenic aggravation, lung cancer with cerebral metastases, and radicular syndrome. CJD and conversion disorder also share many symptoms (Table).
Correct diagnosis of conversion disorder calls for ruling out neurologic and medical conditions. Ms. M’s upbeat nystagmus prompted aggressive neurologic evaluation. Although horizontal nystagmus has been reported rarely in conversion disorder,5 vertical nystagmus has not. One case report6 describes vertical nystagmus as the first clinical sign of CJD.
Leading clinical symptoms of CJD include progressive dementia, myoclonus, cerebellar ataxia, visual problems, and extrapyramidal signs.7 Ms. M’s uncontrollable movements and jerks, although not classically myoclonic, were similar to this common finding. She did not present with dementia, but her rapidly progressive end-stage mental status changes were characteristic of CJD.
Sporadic CJD accounts for 84% of transmissible spongiform encephalopathies. Genetic, iatrogenic, and variant CJD forms (linked to bovine spongiform encephalopathy, or “mad-cow disease”) account for other cases.8 Psychiatric symptoms are a more-common manifestation of variant CJD9 but have been reported in sporadic CJD.10
Eventually, Ms. M’s upbeat nystagmus, persistent abnormal movements, rapidly progressive dementia, and elevated CSF protein 14-3-3 made the CJD diagnosis. Protein 14-3-3 is 94% sensitive and 84% specific for diagnosing CJD.11 Ms. M’s EEG findings did not suggest CJD, but these findings are less sensitive and occur later than the CSF findings.11
Finally, conversion disorder is almost always acute, not slowly progressive as with Ms. M.
Table
Conversion disorder, sporadic Creutzfeldt-Jakob disease share many symptoms
| Complaint | Conversion disorder | Sporadic CJD |
|---|---|---|
| Paralysis | May not follow motor pathways | No |
| Myoclonus | Yes | Cardinal manifestation |
| Ataxia | May be bizarre in character | Present in 25% to 30% of patients, reflecting multiple disease subtypes |
| Hyperreflexia | No | Yes (40% to 80% of patients) |
| Dysphagia | Yes | No |
| Vomiting | Yes | No |
| Aphonia | Yes | No |
| Diplopia | Yes | Rare |
| Nystagmus | Rare | Yes |
| Blindness | Hysterical blindness detectable by ophthalmologic examination | Rare |
| Deafness | Yes | Rare |
| Anesthesia | Yes | No |
| Paresthesia | Yes | No |
| Depression | Yes | Yes |
| Other psychiatry diagnoses | Yes | More common in variant CJD |
| Progressive dementia | No | Cardinal manifestation |
| Temporal relationship with stress | Yes | No |
| Left-side symptoms more common | Yes | No |
Getting the diagnosis right
DSM-IV-TR criteria state that conversion disorder symptoms cannot be otherwise explained “after appropriate investigation,” but what constitutes “appropriate” is unclear. Extensive inpatient evaluation eventually produced the correct diagnosis for Ms. M, but such a detailed evaluation may be too expensive and expansive for every patient with conversion disorder symptoms.
In the conversion disorder study,4 the 10 misdiagnosed patients received one to eight supplemental diagnostic techniques before being correctly diagnosed. In five of the patients, however, the general neurologic examination was identified as the diagnostic technique responsible for final diagnosis.
These findings suggest that a neurologic examination is key to evaluating complaints that suggest conversion disorder and to identify neurologic conditions. The results can also suggest somatic disorders, as exam findings will reflect patients’ perceptions of neurologic processes. For example:
- patients with conversion motor symptoms may have tonic contractures of antagonistic muscles to “paralyze” certain joints
- those with conversion sensory symptoms rarely have sensory impairments that follow known innervation patterns.
Motor complaints, such as localized paralysis or abnormal movements, should be evaluated with a brain MRI to look for lesions along the motor or cerebellar tracts. Sensory pathways can be further investigated with brain MRI and the relevant evoked potential(s) (visual, brainstem, or somatosensory).
Order EEG for patients with convulsions, particularly prolonged EEG monitoring with a video component, and measure serum prolactin immediately after an episode. In some cases, the neurologic exam alone or in conjunction with these initial studies can make the diagnosis. If the clinical situation warrants, more-detailed evaluations may be necessary.
Related resources
- Wise MG, Rundell JR. Clinical manual of psychosomatic medicine: a guide to consultation-liaison psychiatry. Arlington, VA: American Psychiatric Publishing; 2005.
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Duloxetine • Cymbalta
- Mirtazapine • Remeron
The authors report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
History: 3 ‘Uncontrollable’ months
Ms. M, age 57, presents to the ER complaining of coordination problems and involuntary limb movements that have gradually worsened over 3 months.
Two months ago, Ms. M’s primary care physician and neurologist diagnosed her with conversion disorder. Brain MRI at the time showed mild chronic ischemic changes; cervical spinal cord MRI was normal. The neurologist referred Ms. M to a psychiatrist, who prescribed duloxetine, dosage unknown. She started having suicidal thoughts and trembling after starting the medication, so she stopped taking it after 1 week.
Physical exam shows upbeat nystagmus, inconsistent sensory findings, limb ataxia that is more pronounced on the right side, and uncontrollable limb movements, particularly of the right arm.
Ms. M is divorced, lives alone, and works as a medical secretary. Four months ago, she marked the fifth anniversary of her daughter’s death from a drug overdose at age 20. Her parents, whom she cared for, died within the last 3 years. Her son recently left home to attend graduate school, and she is estranged from the rest of her family. She endorses depressed mood and grief over her daughter’s death but says she has no one with whom to talk. She also feels persistent guilt, as she was out on a date when her daughter tried to call home shortly before her death.
The limb movements and lack of coordination are increasingly interfering with Ms. M’s life. She often uses her left hand to stop the right from moving and to guide it in simple tasks, such as opening doors. She can no longer hold a cup of coffee in her right hand or stand on stools at work to reach overhead shelves. At presentation, Ms. M’s imbalance and involuntary movements are so severe that she cannot walk. A coworker drove her to the ER.
poll here
The authors’ observations
A neurologist who evaluates Ms. M in the ER is concerned about her vertical nystagmus, which, unlike horizontal nystagmus, is almost always pathologic. The neurology service admits her for further evaluation.
Ms. M’s age, recent normal MRIs, physical presentation, and lack of other findings suggest a paraneoplastic syndrome. Ataxia associated with subacute cerebellar degeneration can indicate an occult malignancy and is closely linked to gynecologic and breast cancers. Cerebellar degeneration often begins with loss of coordination, can be unilateral, and can appear as intention myoclonus.1
Also considered are:
- opsoclonus-myoclonus, which presents with ataxia, myoclonus, and random chaotic eye movements. This paraneoplastic disorder is less common in adults than in children, however.1
- alien hand/limb syndrome, in which the limb unintentionally performs seemingly purposeful movements, often prompting the patient to restrain the limb with the other hand. This syndrome, however, usually localizes to a lesion in the medial frontal lobe or corpus callosum. Ms. M’s brain MRIs show no such lesion.
poll here
Treatment: Searching for answers
We order an extensive neurologic workup for Ms. M, focusing on causes of inherited and acquired ataxias. The evaluation includes:
- brain and cervical spine MRIs to check for focal cerebral and spinal lesions
- EEG to search for seizure activity and slowing characteristic of encephalopathies
- urine heavy metal testing for toxic processes
- thyroid-stimulating hormone testing for hypothyroid-associated ataxia.
Paraneoplastic workup includes chest, pelvic, and abdominal CT; a gynecologic exam; and a mammogram. All results are negative or equivocal.
We also order blood tests for paraneoplastic antibodies, evidence of opsoclonus/myoclonus, and spinocerebellar ataxia genetic testing; and a CSF check for protein 14-3-3 levels suggestive of prion disease. These tests, run at specialized laboratories, take 4 to 6 weeks.
Ms. M remains hospitalized for 7 days for evaluation. Her movement problems persist, though they often abate when she is distracted. Her upbeat nystagmus appears intermittent. Her affect is diverse, often shifting between tearfulness and inappropriate laughter.
Based on interviews with Ms. M, the C/L team sees prominent depressive symptoms including marked difficulty sleeping, appetite loss, and excessive guilt over her daughter’s death. She also seems indifferent towards her disabling motor symptoms.
The C/L team diagnoses Ms. M with chronic and acute adjustment disorder and major depressive disorder. She is initially hesitant to take another antidepressant but agrees to try mirtazapine, 15 mg nightly, to treat her depression, decreased appetite, and sleep problems. After 2 days, mirtazapine is increased to 30 mg nightly as she is tolerating it and is willing to try a higher dosage.
poll here
The authors’ observations
No neurologic or pathologic explanation is found for Ms. M’s symptoms. Imaging reveals no lesions to explain her intermittent upbeat nystagmus, which localizes to the pons and caudal medulla.2
Conversion disorder. Ms. M, however, appears to meet DSM-IV-TR criteria for conversion disorder (Box), which is thought to result from intense psychological distress in persons who can only express such emotions somatically. Her complaints had specific precursors: she was newly separated from her son and had marked the anniversary of her daughter’s death, which intensified her persistent mourning. We link both circumstances temporally to symptom onset. Also, lack of interest in her serious motor symptoms could be the “la belle indifference” typical of conversion disorder.
Ms. M, however, appears highly suggestible. Her physical symptoms improve soon after her attending psychiatrist suggests that treating her depression will decrease her movements. The neurologists also notice day-to-day fluctuations in her gait disturbance and jerking movements. Distraction techniques produce objective improvement in both symptoms.
- One or more symptoms or deficits affecting voluntary motor or sensory function that suggest a neurological or other general medical condition.
- Psychological factors are judged to be associated with the symptom or deficit because the initiation or exacerbation of the symptom or deficit is preceded by conflicts or other stressors.
- The symptoms or deficit is not intentionally produced or feigned (as in factitious disorder or malingering).
- The symptom or deficit cannot, after appropriate investigation, be fully explained by a general medical condition, or by the direct effects of a substance, or as a culturally sanctioned behavior or experience.
- The symptom or deficit causes clinically significant distress or impairment in social, occupational, or other important areas of functioning or warrants medical evaluation.
- The symptom or deficit is not limited to pain or sexual dysfunction, does not occur exclusively during the course of somatization disorder, and is not better accounted for by another mental disorder.
Specify type of symptom or deficit:
With motor symptom or deficit
With sensory symptom or deficit
With seizures or convulsions
With mixed presentation
Source: Diagnostic and statistical manual of mental disorders (4th ed-text rev). Copyright 2000.
American Psychiatric Association. Reprinted with permission.
In malingering, the patient seeks external incentives for feigned behavior. The role of secondary gain must be considered, as Ms. M’s illness has reunited her with her son, who visits her regularly at the hospital.
Ms. M’s evaluation, however, uncovers no evidence that she is intentionally producing symptoms.
Follow-up: The answer becomes clear
One month after discharge to inpatient rehabilitation, Ms. M is readmitted to the neurology unit. Her uncontrollable limb jerks and ataxia are worse, and she appears demented and near mute. At that time, we learn that the CSF sample sent during her first admission is positive for protein 14-3-3.
Ms. M is diagnosed with Creutzfeldt-Jakob disease (CJD), a spongiform encephalopathy secondary to prion disease. She dies 6 days later. Sporadic CJD is confirmed at autopsy.
The authors’ observations
The literature lists no comprehensive differential diagnosis for conversion disorder, probably because presentations are diverse and the symptoms overlap with innumerable neurologic and medical conditions. This is underscored by the broad differential diagnosis for Ms. M’s ataxia.
In a study to identify organic syndromes initially diagnosed as conversion disorder,4 10 of 85 patients (11.8%) were initially misdiagnosed and later found to have dyskinesia, amyotrophic lateral sclerosis, multiple system atrophy, extrapyramidal syndrome, multiple sclerosis, dementia, Parkinson’s disease with psychogenic aggravation, lung cancer with cerebral metastases, and radicular syndrome. CJD and conversion disorder also share many symptoms (Table).
Correct diagnosis of conversion disorder calls for ruling out neurologic and medical conditions. Ms. M’s upbeat nystagmus prompted aggressive neurologic evaluation. Although horizontal nystagmus has been reported rarely in conversion disorder,5 vertical nystagmus has not. One case report6 describes vertical nystagmus as the first clinical sign of CJD.
Leading clinical symptoms of CJD include progressive dementia, myoclonus, cerebellar ataxia, visual problems, and extrapyramidal signs.7 Ms. M’s uncontrollable movements and jerks, although not classically myoclonic, were similar to this common finding. She did not present with dementia, but her rapidly progressive end-stage mental status changes were characteristic of CJD.
Sporadic CJD accounts for 84% of transmissible spongiform encephalopathies. Genetic, iatrogenic, and variant CJD forms (linked to bovine spongiform encephalopathy, or “mad-cow disease”) account for other cases.8 Psychiatric symptoms are a more-common manifestation of variant CJD9 but have been reported in sporadic CJD.10
Eventually, Ms. M’s upbeat nystagmus, persistent abnormal movements, rapidly progressive dementia, and elevated CSF protein 14-3-3 made the CJD diagnosis. Protein 14-3-3 is 94% sensitive and 84% specific for diagnosing CJD.11 Ms. M’s EEG findings did not suggest CJD, but these findings are less sensitive and occur later than the CSF findings.11
Finally, conversion disorder is almost always acute, not slowly progressive as with Ms. M.
Table
Conversion disorder, sporadic Creutzfeldt-Jakob disease share many symptoms
| Complaint | Conversion disorder | Sporadic CJD |
|---|---|---|
| Paralysis | May not follow motor pathways | No |
| Myoclonus | Yes | Cardinal manifestation |
| Ataxia | May be bizarre in character | Present in 25% to 30% of patients, reflecting multiple disease subtypes |
| Hyperreflexia | No | Yes (40% to 80% of patients) |
| Dysphagia | Yes | No |
| Vomiting | Yes | No |
| Aphonia | Yes | No |
| Diplopia | Yes | Rare |
| Nystagmus | Rare | Yes |
| Blindness | Hysterical blindness detectable by ophthalmologic examination | Rare |
| Deafness | Yes | Rare |
| Anesthesia | Yes | No |
| Paresthesia | Yes | No |
| Depression | Yes | Yes |
| Other psychiatry diagnoses | Yes | More common in variant CJD |
| Progressive dementia | No | Cardinal manifestation |
| Temporal relationship with stress | Yes | No |
| Left-side symptoms more common | Yes | No |
Getting the diagnosis right
DSM-IV-TR criteria state that conversion disorder symptoms cannot be otherwise explained “after appropriate investigation,” but what constitutes “appropriate” is unclear. Extensive inpatient evaluation eventually produced the correct diagnosis for Ms. M, but such a detailed evaluation may be too expensive and expansive for every patient with conversion disorder symptoms.
In the conversion disorder study,4 the 10 misdiagnosed patients received one to eight supplemental diagnostic techniques before being correctly diagnosed. In five of the patients, however, the general neurologic examination was identified as the diagnostic technique responsible for final diagnosis.
These findings suggest that a neurologic examination is key to evaluating complaints that suggest conversion disorder and to identify neurologic conditions. The results can also suggest somatic disorders, as exam findings will reflect patients’ perceptions of neurologic processes. For example:
- patients with conversion motor symptoms may have tonic contractures of antagonistic muscles to “paralyze” certain joints
- those with conversion sensory symptoms rarely have sensory impairments that follow known innervation patterns.
Motor complaints, such as localized paralysis or abnormal movements, should be evaluated with a brain MRI to look for lesions along the motor or cerebellar tracts. Sensory pathways can be further investigated with brain MRI and the relevant evoked potential(s) (visual, brainstem, or somatosensory).
Order EEG for patients with convulsions, particularly prolonged EEG monitoring with a video component, and measure serum prolactin immediately after an episode. In some cases, the neurologic exam alone or in conjunction with these initial studies can make the diagnosis. If the clinical situation warrants, more-detailed evaluations may be necessary.
Related resources
- Wise MG, Rundell JR. Clinical manual of psychosomatic medicine: a guide to consultation-liaison psychiatry. Arlington, VA: American Psychiatric Publishing; 2005.
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Duloxetine • Cymbalta
- Mirtazapine • Remeron
The authors report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Dropcho EJ, Dalmau J, Greenlee JE, et al. Paraneoplastic disorders: Central nervous system disorders. Continuum 1999;5:25-40.
2. Zingler VC, Strupp M, Jahn K, et al. Upbeat nystagmus as the initial clinical sign of Creutzfeldt-Jakob disease. Ann Neurol 2005;57:607-8.
3. Phillips KA (ed). Somatoform and factitious disorders. Washington, DC: American Psychiatric Publishing; 2001.
4. Moene FC, Landberg EH, Hoogduin KA, et al. Organic syndromes diagnosed as conversion disorder: identification and frequency in a study of 85 patients. J Psychosom Res 2000;49:7-12.
5. Smith CH, Beck RW, Mills RP. Functional disease in neuroophthalmology. Neurol Clin 1983;1:955-71.
6. Pierrot-Deseilligny C, Milea D. Vertical nystagmus: clinical facts and hypotheses. Brain 2005;128(Pt 6):1237-46.
7. Glatzel M, Stoeck K, Seeger H, et al. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005;62:545-52.
8. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005;64:1586-91.
9. Spencer MD, Knight RSG, Will RG. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ 2002;324:1479-82.
10. Jiang TT, Moses H, Gordon H, Obah E. Sporadic Creuztfeldt-Jakob disease presenting as major depression. South Med J 1999;92:807-8.
11. Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:811-15.
1. Dropcho EJ, Dalmau J, Greenlee JE, et al. Paraneoplastic disorders: Central nervous system disorders. Continuum 1999;5:25-40.
2. Zingler VC, Strupp M, Jahn K, et al. Upbeat nystagmus as the initial clinical sign of Creutzfeldt-Jakob disease. Ann Neurol 2005;57:607-8.
3. Phillips KA (ed). Somatoform and factitious disorders. Washington, DC: American Psychiatric Publishing; 2001.
4. Moene FC, Landberg EH, Hoogduin KA, et al. Organic syndromes diagnosed as conversion disorder: identification and frequency in a study of 85 patients. J Psychosom Res 2000;49:7-12.
5. Smith CH, Beck RW, Mills RP. Functional disease in neuroophthalmology. Neurol Clin 1983;1:955-71.
6. Pierrot-Deseilligny C, Milea D. Vertical nystagmus: clinical facts and hypotheses. Brain 2005;128(Pt 6):1237-46.
7. Glatzel M, Stoeck K, Seeger H, et al. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005;62:545-52.
8. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005;64:1586-91.
9. Spencer MD, Knight RSG, Will RG. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ 2002;324:1479-82.
10. Jiang TT, Moses H, Gordon H, Obah E. Sporadic Creuztfeldt-Jakob disease presenting as major depression. South Med J 1999;92:807-8.
11. Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:811-15.