An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
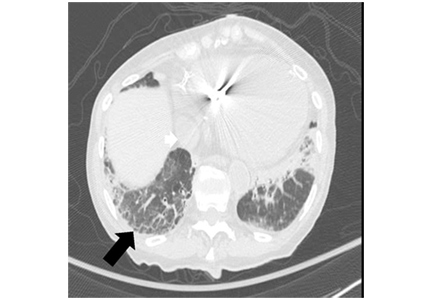
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540
An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540