User login
Case Report
Hyperhemolysis syndrome is a form of atypical hemolytic transfusion reaction (HTR). It is characterized by a significant drop in hemoglobin (Hb) after seemingly compatible red blood cell transfusions, suggesting destruction of both transfused and autologous red blood cells. Its pathophysiology is not well understood, and a serologic cause is often not identified.14 In contrast, delayed HTRs are typically characterized by a positive direct antiglobulin test (DAT), suggesting that the patient's red blood cells are coated by immunoglobulin G and/or complement components and by the appearance of previously undetected red blood cell alloantibody or antibodies that developed from a secondary anamnestic response; however, autologous red cells are not destroyed.
CASE
A 48‐year‐old African American woman with sickle cell disease (SCD) was readmitted for pain crisis. Her medical history included stroke, pulmonary hypertension, and congestive heart failure. She had received several transfusions and consequently had developed antibodies to seven clinically significant red blood cell antigens. A week prior to readmission, she was discharged from the hospital with an Hb of 6.9 g/dL after a sickle cell crisis precipitated by pneumonia. She was treated with hydration, pain medications, antibiotics, and a unit of cross‐match‐compatible red blood cells (RBCs) that was antigen negative for her antibodies.
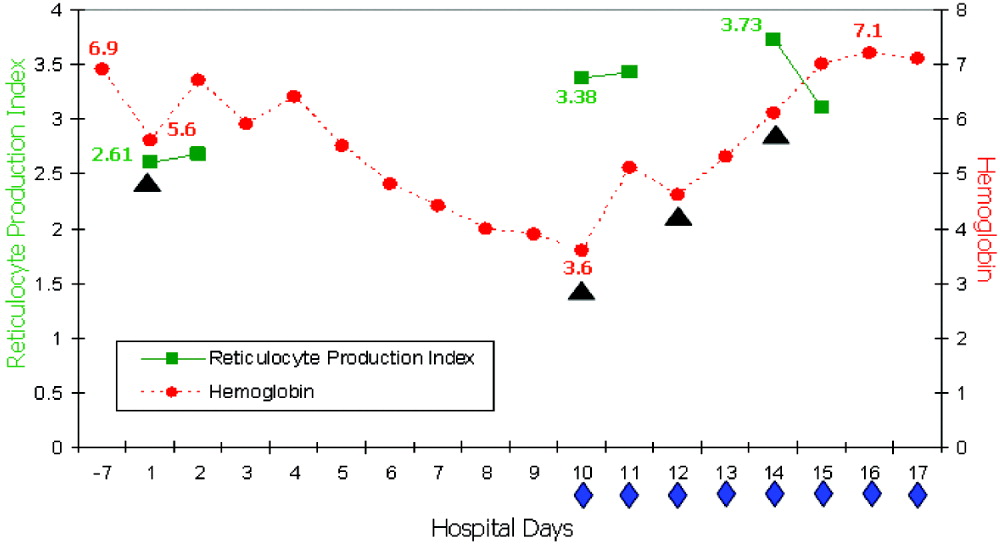
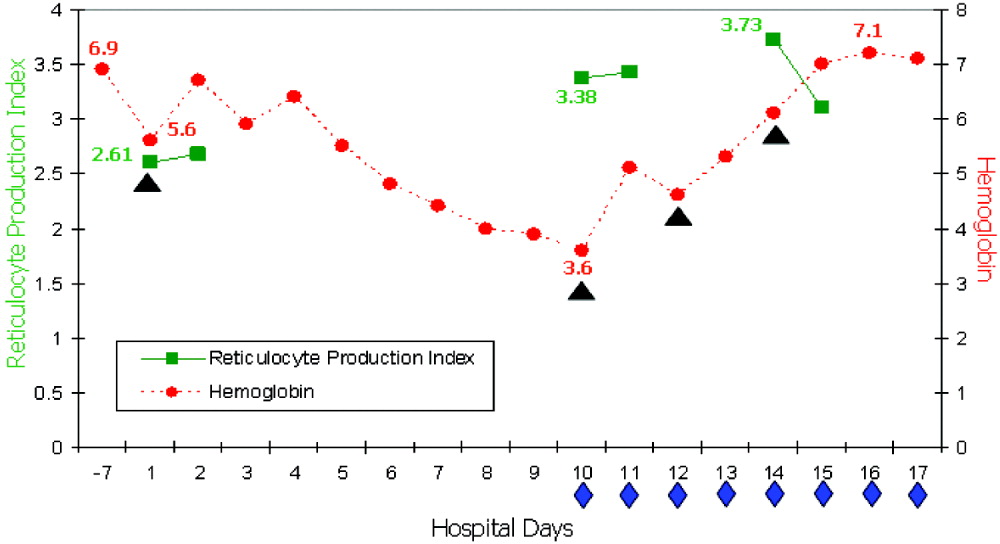
On readmission, she had an Hb of 5.6 g/dL and an uncorrected reticulocyte count of 17.6%. Her reticulocyte production index, a reticulocyte count corrected for the degree of anemia and reticulocyte maturation time, was elevated at 2.6. She was transfused with 1 unit of phenotypically matched and cross‐match‐compatible RBCs. Three hours after transfusion, she developed dark‐colored urine. The transfusion reaction investigation revealed no clerical error or incompatibility, a negative DAT, and an antibody panel identical to that from pretransfusion testing. During hospitalization, the hemolytic anemia worsened (Fig. 1). On the 10th hospital day, she became severely dyspneic as her Hb reached its nadir of 3.6 g/dL despite ongoing erythropoiesis. She developed decompensated heart failure and renal insufficiency, precipitated by the acutely worsening anemia. Along with diuretic and vasodilator therapies, she was treated with methylprednisolone at 125 mg twice daily for 3 days followed by tapering doses of prednisone for 2 weeks, intravenous immunoglobulin (IVIG) at 400 mg/kg a day for 5 days, and 4 cross‐match‐compatible RBC transfusions that were antigen negative for her antibodies. The hemolysis resolved and the patient improved. Throughout hospitalization, her DAT remained negative. The Hb remained stable at 7 g/dL until she was discharged. Ten months of follow‐up showed no new red blood cell antibody in her serum or recurrence of hyperhemolysis syndrome despite receiving subsequent transfusions.
DISCUSSION
Hyperhemolysis syndrome has been described in patients with SCD,14, 6, 7 suggesting that an underlying hemoglobinopathy may be a risk factor; however, a patient with anemia of chronic disease was recently described in the literature to have developed hyperhemolysis syndrome.5 Possible mechanisms include innocent bystander hemolysis through complement‐mediated lysis and/or formation of red blood cell alloantibody or autoantibody;1, 2 and hyperactive macrophages of the reticuloendothelial system that recognize Hb S RBCs of patients with SCD more avidly than normal RBCs because of the exposure of aminophosphatides in the outer layer of the sickled RBC membrane.3 In effect, red blood cells may be destroyed regardless of whether they are autologous or transfused. Additionally, transfusion‐related suppression of erythropoiesis may worsen the severity of anemia.2 Recent studies of patients with SCD suggest that the presence of free plasma Hb, as a consequence of hemolysis, reduces nitric oxide bioavailability, promotes endothelial dysfunction, and contributes to the development of pulmonary hypertension and the varying presentations of vasoocclusion.6 A common observation among patients who experience hyperhemolysis syndrome is that withholding transfusion seems beneficial, probably because immunologic reactions are not exacerbated, whereas treatment with steroids1, 2, 4 and/or IVIG3, 7 resolves hemolysis because of their immunomodulatory effects.
CONCLUSIONS
Hyperhemolysis syndrome is a potentially life‐threatening complication of RBC transfusion. It is important to recognize this syndrome when managing patients with SCD who present with worsening anemia after RBC transfusions. Although further transfusions can exacerbate hemolysis4, 7 and may be relatively contraindicated, in severe and desperate situations, simultaneous treatment with steroids and IVIG, together with RBC transfusions, may be lifesaving.
- ,,,,.Delayed hemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients' red cells.Transfusion.1997;37:376–381.
- ,,,,.The sickle cell hemolytic transfusion reaction syndrome.Transfusion.1997;37:382–392.
- ,,,,.Hyperhemolytic transfusion reaction in sickle cell disease.Transfusion.2001;41:323–328.
- ,,,,.Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease.Pediatrics.2003;111(6 Pt 1):e661–e665.
- ,.Hyperhemolysis syndrome in anemia of chronic disease.Transfusion.2005;45:1930–1933.
- and.Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia.Transfusion.2006;46:105–110.
- ,,,.Post‐transfusion hyperhemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction.Vox Sang.1995;69:355–357.
Hyperhemolysis syndrome is a form of atypical hemolytic transfusion reaction (HTR). It is characterized by a significant drop in hemoglobin (Hb) after seemingly compatible red blood cell transfusions, suggesting destruction of both transfused and autologous red blood cells. Its pathophysiology is not well understood, and a serologic cause is often not identified.14 In contrast, delayed HTRs are typically characterized by a positive direct antiglobulin test (DAT), suggesting that the patient's red blood cells are coated by immunoglobulin G and/or complement components and by the appearance of previously undetected red blood cell alloantibody or antibodies that developed from a secondary anamnestic response; however, autologous red cells are not destroyed.
CASE
A 48‐year‐old African American woman with sickle cell disease (SCD) was readmitted for pain crisis. Her medical history included stroke, pulmonary hypertension, and congestive heart failure. She had received several transfusions and consequently had developed antibodies to seven clinically significant red blood cell antigens. A week prior to readmission, she was discharged from the hospital with an Hb of 6.9 g/dL after a sickle cell crisis precipitated by pneumonia. She was treated with hydration, pain medications, antibiotics, and a unit of cross‐match‐compatible red blood cells (RBCs) that was antigen negative for her antibodies.
On readmission, she had an Hb of 5.6 g/dL and an uncorrected reticulocyte count of 17.6%. Her reticulocyte production index, a reticulocyte count corrected for the degree of anemia and reticulocyte maturation time, was elevated at 2.6. She was transfused with 1 unit of phenotypically matched and cross‐match‐compatible RBCs. Three hours after transfusion, she developed dark‐colored urine. The transfusion reaction investigation revealed no clerical error or incompatibility, a negative DAT, and an antibody panel identical to that from pretransfusion testing. During hospitalization, the hemolytic anemia worsened (Fig. 1). On the 10th hospital day, she became severely dyspneic as her Hb reached its nadir of 3.6 g/dL despite ongoing erythropoiesis. She developed decompensated heart failure and renal insufficiency, precipitated by the acutely worsening anemia. Along with diuretic and vasodilator therapies, she was treated with methylprednisolone at 125 mg twice daily for 3 days followed by tapering doses of prednisone for 2 weeks, intravenous immunoglobulin (IVIG) at 400 mg/kg a day for 5 days, and 4 cross‐match‐compatible RBC transfusions that were antigen negative for her antibodies. The hemolysis resolved and the patient improved. Throughout hospitalization, her DAT remained negative. The Hb remained stable at 7 g/dL until she was discharged. Ten months of follow‐up showed no new red blood cell antibody in her serum or recurrence of hyperhemolysis syndrome despite receiving subsequent transfusions.
DISCUSSION
Hyperhemolysis syndrome has been described in patients with SCD,14, 6, 7 suggesting that an underlying hemoglobinopathy may be a risk factor; however, a patient with anemia of chronic disease was recently described in the literature to have developed hyperhemolysis syndrome.5 Possible mechanisms include innocent bystander hemolysis through complement‐mediated lysis and/or formation of red blood cell alloantibody or autoantibody;1, 2 and hyperactive macrophages of the reticuloendothelial system that recognize Hb S RBCs of patients with SCD more avidly than normal RBCs because of the exposure of aminophosphatides in the outer layer of the sickled RBC membrane.3 In effect, red blood cells may be destroyed regardless of whether they are autologous or transfused. Additionally, transfusion‐related suppression of erythropoiesis may worsen the severity of anemia.2 Recent studies of patients with SCD suggest that the presence of free plasma Hb, as a consequence of hemolysis, reduces nitric oxide bioavailability, promotes endothelial dysfunction, and contributes to the development of pulmonary hypertension and the varying presentations of vasoocclusion.6 A common observation among patients who experience hyperhemolysis syndrome is that withholding transfusion seems beneficial, probably because immunologic reactions are not exacerbated, whereas treatment with steroids1, 2, 4 and/or IVIG3, 7 resolves hemolysis because of their immunomodulatory effects.
CONCLUSIONS
Hyperhemolysis syndrome is a potentially life‐threatening complication of RBC transfusion. It is important to recognize this syndrome when managing patients with SCD who present with worsening anemia after RBC transfusions. Although further transfusions can exacerbate hemolysis4, 7 and may be relatively contraindicated, in severe and desperate situations, simultaneous treatment with steroids and IVIG, together with RBC transfusions, may be lifesaving.
Hyperhemolysis syndrome is a form of atypical hemolytic transfusion reaction (HTR). It is characterized by a significant drop in hemoglobin (Hb) after seemingly compatible red blood cell transfusions, suggesting destruction of both transfused and autologous red blood cells. Its pathophysiology is not well understood, and a serologic cause is often not identified.14 In contrast, delayed HTRs are typically characterized by a positive direct antiglobulin test (DAT), suggesting that the patient's red blood cells are coated by immunoglobulin G and/or complement components and by the appearance of previously undetected red blood cell alloantibody or antibodies that developed from a secondary anamnestic response; however, autologous red cells are not destroyed.
CASE
A 48‐year‐old African American woman with sickle cell disease (SCD) was readmitted for pain crisis. Her medical history included stroke, pulmonary hypertension, and congestive heart failure. She had received several transfusions and consequently had developed antibodies to seven clinically significant red blood cell antigens. A week prior to readmission, she was discharged from the hospital with an Hb of 6.9 g/dL after a sickle cell crisis precipitated by pneumonia. She was treated with hydration, pain medications, antibiotics, and a unit of cross‐match‐compatible red blood cells (RBCs) that was antigen negative for her antibodies.
On readmission, she had an Hb of 5.6 g/dL and an uncorrected reticulocyte count of 17.6%. Her reticulocyte production index, a reticulocyte count corrected for the degree of anemia and reticulocyte maturation time, was elevated at 2.6. She was transfused with 1 unit of phenotypically matched and cross‐match‐compatible RBCs. Three hours after transfusion, she developed dark‐colored urine. The transfusion reaction investigation revealed no clerical error or incompatibility, a negative DAT, and an antibody panel identical to that from pretransfusion testing. During hospitalization, the hemolytic anemia worsened (Fig. 1). On the 10th hospital day, she became severely dyspneic as her Hb reached its nadir of 3.6 g/dL despite ongoing erythropoiesis. She developed decompensated heart failure and renal insufficiency, precipitated by the acutely worsening anemia. Along with diuretic and vasodilator therapies, she was treated with methylprednisolone at 125 mg twice daily for 3 days followed by tapering doses of prednisone for 2 weeks, intravenous immunoglobulin (IVIG) at 400 mg/kg a day for 5 days, and 4 cross‐match‐compatible RBC transfusions that were antigen negative for her antibodies. The hemolysis resolved and the patient improved. Throughout hospitalization, her DAT remained negative. The Hb remained stable at 7 g/dL until she was discharged. Ten months of follow‐up showed no new red blood cell antibody in her serum or recurrence of hyperhemolysis syndrome despite receiving subsequent transfusions.
DISCUSSION
Hyperhemolysis syndrome has been described in patients with SCD,14, 6, 7 suggesting that an underlying hemoglobinopathy may be a risk factor; however, a patient with anemia of chronic disease was recently described in the literature to have developed hyperhemolysis syndrome.5 Possible mechanisms include innocent bystander hemolysis through complement‐mediated lysis and/or formation of red blood cell alloantibody or autoantibody;1, 2 and hyperactive macrophages of the reticuloendothelial system that recognize Hb S RBCs of patients with SCD more avidly than normal RBCs because of the exposure of aminophosphatides in the outer layer of the sickled RBC membrane.3 In effect, red blood cells may be destroyed regardless of whether they are autologous or transfused. Additionally, transfusion‐related suppression of erythropoiesis may worsen the severity of anemia.2 Recent studies of patients with SCD suggest that the presence of free plasma Hb, as a consequence of hemolysis, reduces nitric oxide bioavailability, promotes endothelial dysfunction, and contributes to the development of pulmonary hypertension and the varying presentations of vasoocclusion.6 A common observation among patients who experience hyperhemolysis syndrome is that withholding transfusion seems beneficial, probably because immunologic reactions are not exacerbated, whereas treatment with steroids1, 2, 4 and/or IVIG3, 7 resolves hemolysis because of their immunomodulatory effects.
CONCLUSIONS
Hyperhemolysis syndrome is a potentially life‐threatening complication of RBC transfusion. It is important to recognize this syndrome when managing patients with SCD who present with worsening anemia after RBC transfusions. Although further transfusions can exacerbate hemolysis4, 7 and may be relatively contraindicated, in severe and desperate situations, simultaneous treatment with steroids and IVIG, together with RBC transfusions, may be lifesaving.
- ,,,,.Delayed hemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients' red cells.Transfusion.1997;37:376–381.
- ,,,,.The sickle cell hemolytic transfusion reaction syndrome.Transfusion.1997;37:382–392.
- ,,,,.Hyperhemolytic transfusion reaction in sickle cell disease.Transfusion.2001;41:323–328.
- ,,,,.Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease.Pediatrics.2003;111(6 Pt 1):e661–e665.
- ,.Hyperhemolysis syndrome in anemia of chronic disease.Transfusion.2005;45:1930–1933.
- and.Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia.Transfusion.2006;46:105–110.
- ,,,.Post‐transfusion hyperhemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction.Vox Sang.1995;69:355–357.
- ,,,,.Delayed hemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients' red cells.Transfusion.1997;37:376–381.
- ,,,,.The sickle cell hemolytic transfusion reaction syndrome.Transfusion.1997;37:382–392.
- ,,,,.Hyperhemolytic transfusion reaction in sickle cell disease.Transfusion.2001;41:323–328.
- ,,,,.Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease.Pediatrics.2003;111(6 Pt 1):e661–e665.
- ,.Hyperhemolysis syndrome in anemia of chronic disease.Transfusion.2005;45:1930–1933.
- and.Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia.Transfusion.2006;46:105–110.
- ,,,.Post‐transfusion hyperhemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction.Vox Sang.1995;69:355–357.