User login
How Should Hospitalized Patients with Long QT Syndrome Be Managed?
Case
You are asked to admit a 63-year-old male with a history of hypertension and osteoarthritis. The patient, who fell at home, is scheduled for open repair of his femoral neck fracture the following day. The patient reports tripping over his granddaughter’s toys and denies any associated symptoms around the time of his fall. An electrocardiogram (ECG) reveals a QTc (QT) interval of 480 ms. How should this hospitalized patient’s prolonged QT interval be managed?
Overview
Patients with a prolonged QT interval on routine ECG present an interesting dilemma for clinicians. Although QT prolongation—either congenital or acquired—has been associated with dysrhythmias, the risk of torsades de pointes and sudden cardiac death varies considerably based on myriad underlying factors.1 Therefore, the principle job of the clinician who has recognized QT prolongation is to assess and minimize the risk of the development of clinically significant dysrhythmias, and to be prepared to manage them should they arise.
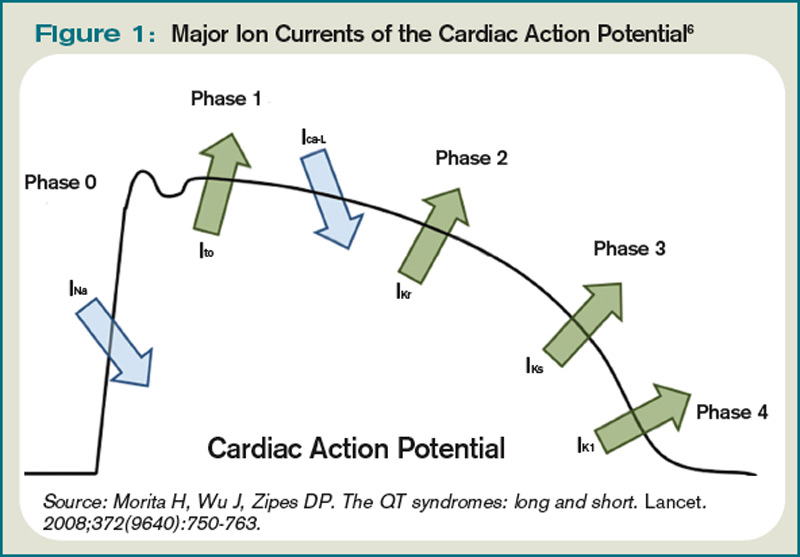
The QT interval encompasses ventricular depolarization and repolarization. This ventricular action potential proceeds through five phases. The initial upstroke (phase 0) of depolarization occurs with the opening of Na+ channels, triggering the inward Na+ current (INa), and causes the interior of the myocytes to become positively charged. This is followed by initial repolarization (phase 1) when the opening of K+ channels causes an outward K+ current (Ito). Next, the plateau phase (phase 2) of the action potential follows with a balance of inward current through Ca2+channels (Ica-L) and outward current through slow rectifier K+ channels (IKs), and then later through delayed, rapid K+ rectifier channels (IKr). Then, the inward current is deactivated, while the outward current increases through the rapid delayed rectifier (IKr) and opening of inward rectifier channels (IK1) to complete repolarization (phase 3). Finally, the action potential returns to baseline (phase 4) and Na+ begins to enter the cell again (see Figure 1, above).
The long QT syndrome (LQTS) is defined by a defect in these cardiac ion channels, which leads to abnormal repolarization, usually lengthening the QT interval and thus predisposing to ventricular dysrhythmias.2 It is estimated that as many as 85% of these syndromes are inherited, and up to 15% are acquired or sporadic.3 Depending on the underlying etiology of the LQTS, manifestations might first be appreciated at any time from in utero through adulthood.4 Symptoms including palpitations, syncope, seizures, or cardiac arrest bring these patients to medical attention.3 These symptoms frequently elicit physical or emotional stress, but they can occur without obvious inciting triggers.5 A 20% mortality risk exists in patients who are symptomatic and untreated in the first year following diagnosis, and up to 50% within 10 years following diagnosis.4
How is Long QT Syndrome Diagnosed?
The LQTS diagnosis is based on clinical history in combination with ECG abnormalities.6 Important historical elements include symptoms of palpitations, syncope, seizures, or cardiac arrest.3 In addition, a family history of unexplained syncope or sudden death, especially at a young age, should raise LQTS suspicion.5
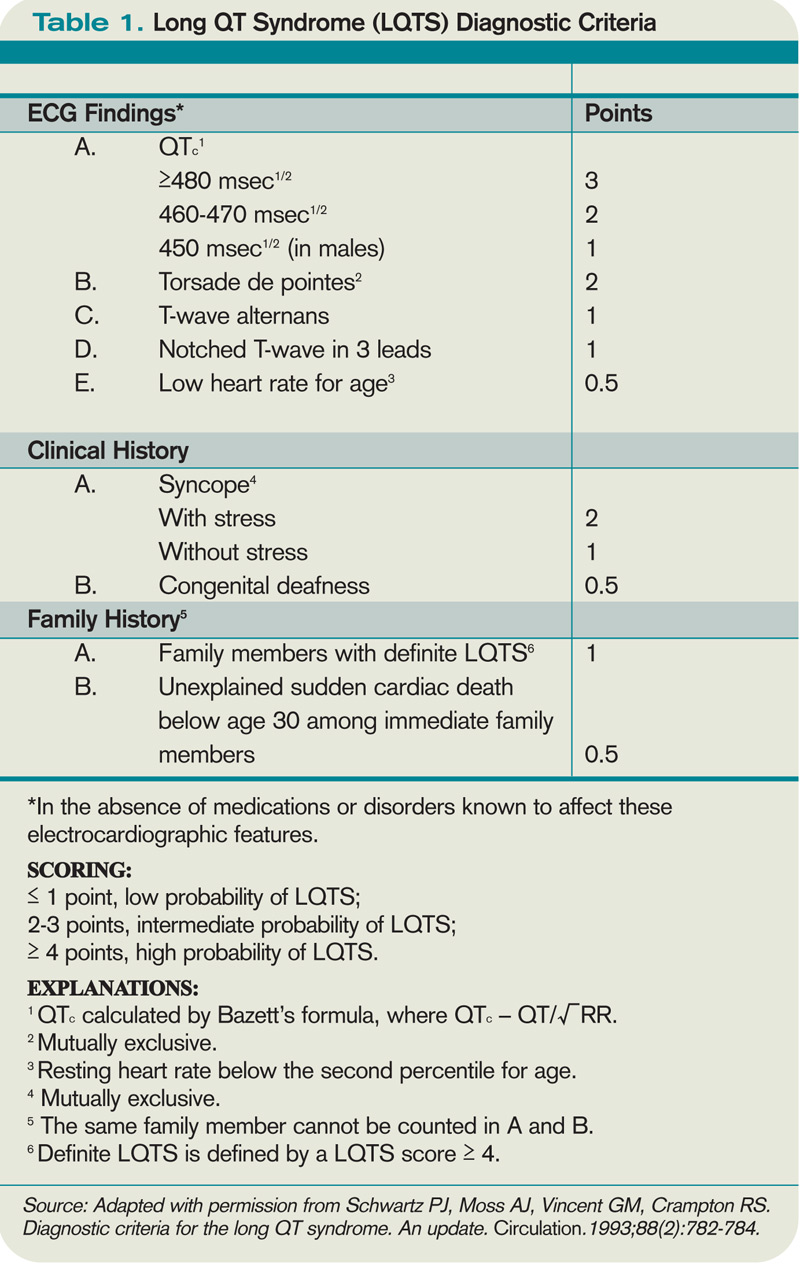
A variety of ECG findings can be witnessed in LQTS patients.4,5 Although the majority of patients have a QTc >440 ms, approximately one-third have a QTc ≤460 ms, and about 10% have normal QTc intervals.5 Other ECG abnormalities include notched, biphasic, or prolonged T-waves, and the presence of U-waves.4,5 Schwartz et al used these elements to publish criteria (see Table 1, right) that physicians can use to assess the probability that a patient has LQTS.7
Types of Long QT Syndromes
Because the risk of developing significant dysrhythmias with LQTS is dependent on both the actual QT interval, with risk for sudden cardiac death increased two to three times with QT >440 ms compared with QT <440 ms and the specific underlying genotype, it is important to have an understanding of congenital and acquired LQTS and the associated triggers for torsades de pointes.
Congenital LQTS
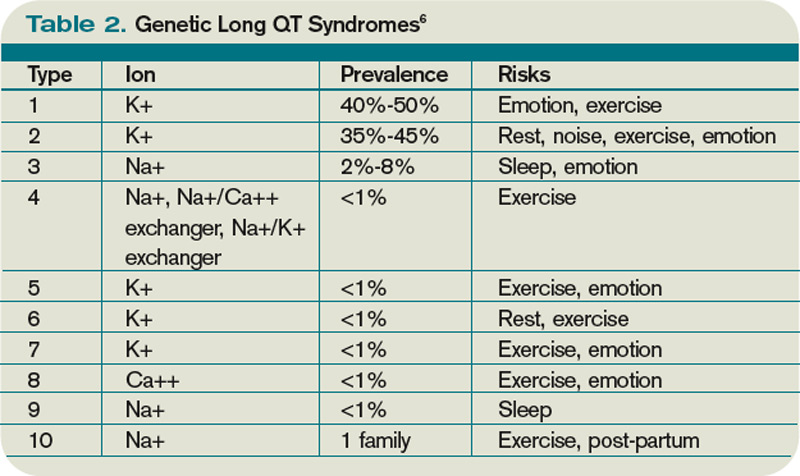
Congenital LQTS is caused by mutations in cardiac ion channel proteins, primarily sodium, and potassium channels.5,6 These defects either slow depolarization or lengthen repolarization, leading to heterogeneity of repolarization of the membrane.5 This, in turn, predisposes to ventricular dysrhythmias, including torsades de pointes and subsequent ventricular fibrillation and death.2 Currently, 12 genetic defects have been identified in LQTS. Hundreds of mutations have been described to cause these defects (see Table 2, right).8 Approximately 70% of congenital LQTS are caused by mutations in three genes and are classified as LQTS 1, LQTS 2, and LQTS 3.8 The other seven mutation types account for about 5% of cases; a quarter of LQTS cases have no identified genetic mutations.8
LQTS usually can be distinguished by clinical features and some ECG characteristics, but diagnosis of the specific type requires genetic testing.8,9 The most common types of LQTS are discussed below.
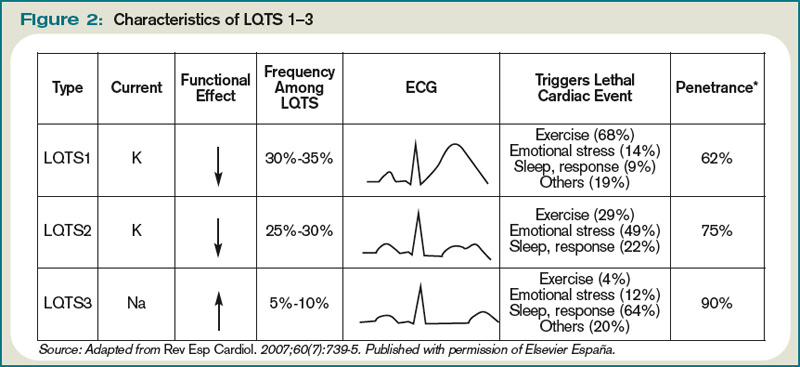
- Long QT1 is the most common type, occurring in approximately 40% to 50% of patients diagnosed with LQTS. It is characterized by a defect in the potassium channel alpha subunit leading to IKs reduction.9 These patients typically present with syncope or adrenergic-induced torsades, might have wide, broad-based T-waves on ECG, and respond well to beta-blocker therapy.6 Triggers for these patients include physical exertion or emotional stressors, particularly exercise and swimming. These patients typically present in early childhood.1
- Long QT2 occurs in 35% to 40% of patients and is characterized by a different defect in the alpha subunit of the potassium channel, which leads to reduced IKr.9 ECGs in LQTS2 can demonstrate low-amplitude and notched T-waves. Sudden catecholamine surges related to emotional stress or loud noises and bradycardia can trigger dysrhythmias in Long QT2.6 Thus, beta blockers reduce overall cardiac events in LQTS2 but less effectively than in LQTS1.6 These patients also present in childhood but typically are older than patients with LQTS1.6
- Long QT3 is less common than LQTS1 or LQTS2, at 2% to 8% of LQTS patients, but carries a higher mortality and is not treated effectively with beta blockers. LQTS3 is characterized by a defect in a sodium channel, causing a gain-of-function in the INa.4,9 These patients are more likely to have a fatal dysrhythmia while sleeping, are less susceptible to exercise-induced events, and have increased morbidity and mortality associated with bradycardia.4,9 ECG frequently reveals a relatively long ST segment, followed by a peaked and tall T-wave. Beta-blocker therapy can predispose to dysrhythmias in these patients; therefore, many of these patients will have pacemakers implanted as first-line therapy.6
While less common, Jervell and Lange Nielson syndrome is an autosomal recessive form of LQTS in which affected patients have homozygous mutations in the KCNQ1 or KCNE1 genes. This syndrome occurs in approximately 1% to 7% of LQTS patients, displays a typical QTc >550 ms, can be triggered by exercise and emotional stress, is associated with deafness, and carries a high risk of cardiac events at a young age.6
Acquired Syndromes
In addition to congenital LQTS, certain patients can acquire LQTS after being treated with particular drugs or having metabolic abnormalities, namely hypomagnesemia, hypocalcemia, and hypokalemia. Most experts think patients who “acquire” LQTS that predisposes to torsades de pointes have underlying structural heart disease or LQTS genetic carrier mutations that combine with transient initiating events (e.g., drugs or metabolic abnormalities) to induce dysrhythmias.1 In addition to certain drugs, cardiac ischemia, and electrolyte abnormalities, cocaine abuse, HIV, and subarachnoid hemorrhage can induce dysrhythmias in susceptible patients.5
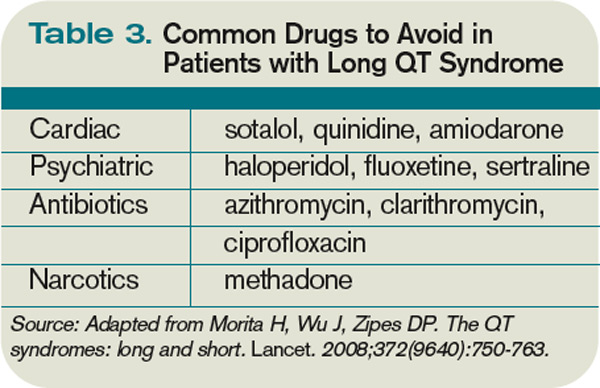
Many types of drugs can cause a prolonged QT interval, and others should be avoided in patients with pre-existing prolonged QT (see Table 3, p. 17). Potentially offending drugs that are frequently encountered by inpatient physicians include amiodarone, diltiazem, erythromycin, clarithromycin, ciprofloxacin, fluoxetine, paroxetine, sertraline, haloperidol, ritonavir, and methadone.1 Additionally, drugs that cause electrolyte abnormalities (e.g., diuretics and lithium) should be monitored closely.
Overall, the goals of therapy in LQTS are:
- Decrease the risk of dysrhythmic events;
- Minimize adrenergic response;
- Shorten the QTc;
- Decrease the dispersion of refractoriness; and
- Improve the function of the ion channels.3
Supportive measures should be taken for patients who are acutely symptomatic from LQTS and associated torsades de pointes. In addition to immediate cardioversion for ongoing and hemodynamically significant torsades, intravenous magnesium should be administered, electrolytes corrected, and offending drugs discontinued.5 Temporary transvenous pacing at rates of approximately 100 beats per minute is highly effective in preventing short-term recurrence of torsades in congenital and acquired LQTS, especially in bradycardic patients.5 Isoproterenol infusion increases the heart rate and effectively prevents acute recurrence of torsades in patients with acquired LQTS, but it should be used with caution in patients with structural heart disease.5
Long-term strategies to manage LQTS include:
- Minimizing the risk of triggering cardiac events via adrenergic stimulation;
- Preventing ongoing dysrhythmias;
- Avoiding medications known to prolong the QT interval; and
- Maintaining normal electrolytes and minerals.5
Most patients with congenital long QT should be discouraged from participating in competitive sports, and patients should attempt to eliminate exposures to stress or sudden awakening, though this is not practical in all cases.5 Beta blockers generally are the first-line therapy and are more effective for LQT1 than LQT2 or LQT3.4,5 If patients are still symptomatic despite adequate medical therapy, or have survived cardiac arrest, they should be considered for ICD therapy.4,5 In addition, patients with profound bradycardia benefit from pacemaker implantation.5 Patients who remain symptomatic despite both beta blockade and ICD placement might find cervicothoracic sympathectomy curative.4,5
Perioperative Considerations
Although little data is available to guide physicians in the prevention of torsades de pointes during the course of anesthesia, there are a number of considerations that may reduce the chances of symptomatic dysrhythmias.
First, care should be taken to avoid dysrhythmia triggers in LQTS by providing a calm, quiet environment during induction, monitoring, and treating metabolic abnormalities, and providing an appropriate level of anesthesia.10 Beta-blocker therapy should be continued and potentially measured preoperatively by assessing heart rate response during stress testing.5 An implantable cardioverter-defibrillator (AICD) should be interrogated prior to surgery and inactivated during the operation.5
Finally, Kies et al have recommended general anesthesia with propofol for induction (or throughout), isoflurane as the volatile agent, vecuronium for muscle relaxation, and intravenous fentanyl for analgesia when possible.10
Back to the Case
While the patient had no genetic testing for LQTS, evaluation of previous ECGs demonstrated a prolonged QT interval. The hip fracture repair was considered an urgent procedure, which precluded the ability to undertake genetic testing and consideration for device implantation. The only medication that was known to increase the risk for dysrhythmias in this patient was his diuretic, with the attendant risk of electrolyte abnormalities.
Thus, the patient’s hydrochlorothiazide was discontinued and his pre-existing atenolol continued. The patient’s electrolytes and minerals were monitored closely, and magnesium was administered on the day of surgery. Anesthesia was made aware of the prolonged QT interval, such that they were able to minimize the risk for and anticipate the treatment of dysrhythmias. The patient tolerated the surgery and post-operative period without complication and was scheduled for an outpatient workup and management for his prolonged QT interval.
Bottom Line
Long QT syndrome is frequently genetic in origin, but it can be caused by certain medications and perturbations of electrolytes. Beta blockers are the first-line therapy for the majority of LQTS cases, along with discontinuation of drugs that might induce or aggravate the QT prolongation.
Patients who have had cardiac arrest or who remain symptomatic despite beta-blocker therapy should have an ICD implanted.
In the perioperative period, patients’ electrolytes should be monitored and kept within normal limits. If the patient is on a beta blocker, it should be continued, and the anesthesiologist should be made aware of the diagnosis so that the anesthethic plan can be optimized to prevent arrhythmic complications. TH
Dr. Kamali is a medical resident at the University of Colorado Denver. Dr. Stickrath is a hospitalist at the Veterans Affairs Medical Center in Denver and an instructor of medicine at UC Denver. Dr. Prochazka is director of ambulatory care at the Denver VA and professor of medicine at UC Denver. Dr. Varosy is director of cardiac electrophysiology at the Denver VA and assistant professor of medicine at UC Denver.
References
- Kao LW, Furbee BR. Drug-induced q-T prolongation. Med Clin North Am. 2005;89(6):1125-1144.
- Marchlinski F. Chapter 226, The Tachyarrhythmias; Harrison's Principles of Internal Medicine, 17e. Available at: www.accessmedicine.com/resourceTOC .aspx?resourceID=4. Accessed Nov. 21, 2009.
- Zareba W, Cygankiewicz I. Long QT syndrome and short QT syndrome. Prog Cardiovasc Dis. 2008; 51(3):264-278.
- Booker PD, Whyte SD, Ladusans EJ. Long QT syndrome and anaesthesia. Br J Anaesth. 2003;90(3):349-366.
- Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002;143(1):7-14.
- Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372(9640):750-763.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-784.
- Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752-1760.
- Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med. 2006;8(3):143-155.
- Kies SJ, Pabelick CM, Hurley HA, White RD, Ackerman MJ. Anesthesia for patients with congenital long QT syndrome. Anesthesiology. 2005;102(1):204-210.
- Wisely NA, Shipton EA. Long QT syndrome and anaesthesia. Eur J Anaesthesiol. 2002;19(12):853-859.
Case
You are asked to admit a 63-year-old male with a history of hypertension and osteoarthritis. The patient, who fell at home, is scheduled for open repair of his femoral neck fracture the following day. The patient reports tripping over his granddaughter’s toys and denies any associated symptoms around the time of his fall. An electrocardiogram (ECG) reveals a QTc (QT) interval of 480 ms. How should this hospitalized patient’s prolonged QT interval be managed?
Overview
Patients with a prolonged QT interval on routine ECG present an interesting dilemma for clinicians. Although QT prolongation—either congenital or acquired—has been associated with dysrhythmias, the risk of torsades de pointes and sudden cardiac death varies considerably based on myriad underlying factors.1 Therefore, the principle job of the clinician who has recognized QT prolongation is to assess and minimize the risk of the development of clinically significant dysrhythmias, and to be prepared to manage them should they arise.
The QT interval encompasses ventricular depolarization and repolarization. This ventricular action potential proceeds through five phases. The initial upstroke (phase 0) of depolarization occurs with the opening of Na+ channels, triggering the inward Na+ current (INa), and causes the interior of the myocytes to become positively charged. This is followed by initial repolarization (phase 1) when the opening of K+ channels causes an outward K+ current (Ito). Next, the plateau phase (phase 2) of the action potential follows with a balance of inward current through Ca2+channels (Ica-L) and outward current through slow rectifier K+ channels (IKs), and then later through delayed, rapid K+ rectifier channels (IKr). Then, the inward current is deactivated, while the outward current increases through the rapid delayed rectifier (IKr) and opening of inward rectifier channels (IK1) to complete repolarization (phase 3). Finally, the action potential returns to baseline (phase 4) and Na+ begins to enter the cell again (see Figure 1, above).
The long QT syndrome (LQTS) is defined by a defect in these cardiac ion channels, which leads to abnormal repolarization, usually lengthening the QT interval and thus predisposing to ventricular dysrhythmias.2 It is estimated that as many as 85% of these syndromes are inherited, and up to 15% are acquired or sporadic.3 Depending on the underlying etiology of the LQTS, manifestations might first be appreciated at any time from in utero through adulthood.4 Symptoms including palpitations, syncope, seizures, or cardiac arrest bring these patients to medical attention.3 These symptoms frequently elicit physical or emotional stress, but they can occur without obvious inciting triggers.5 A 20% mortality risk exists in patients who are symptomatic and untreated in the first year following diagnosis, and up to 50% within 10 years following diagnosis.4
How is Long QT Syndrome Diagnosed?
The LQTS diagnosis is based on clinical history in combination with ECG abnormalities.6 Important historical elements include symptoms of palpitations, syncope, seizures, or cardiac arrest.3 In addition, a family history of unexplained syncope or sudden death, especially at a young age, should raise LQTS suspicion.5
A variety of ECG findings can be witnessed in LQTS patients.4,5 Although the majority of patients have a QTc >440 ms, approximately one-third have a QTc ≤460 ms, and about 10% have normal QTc intervals.5 Other ECG abnormalities include notched, biphasic, or prolonged T-waves, and the presence of U-waves.4,5 Schwartz et al used these elements to publish criteria (see Table 1, right) that physicians can use to assess the probability that a patient has LQTS.7
Types of Long QT Syndromes
Because the risk of developing significant dysrhythmias with LQTS is dependent on both the actual QT interval, with risk for sudden cardiac death increased two to three times with QT >440 ms compared with QT <440 ms and the specific underlying genotype, it is important to have an understanding of congenital and acquired LQTS and the associated triggers for torsades de pointes.
Congenital LQTS
Congenital LQTS is caused by mutations in cardiac ion channel proteins, primarily sodium, and potassium channels.5,6 These defects either slow depolarization or lengthen repolarization, leading to heterogeneity of repolarization of the membrane.5 This, in turn, predisposes to ventricular dysrhythmias, including torsades de pointes and subsequent ventricular fibrillation and death.2 Currently, 12 genetic defects have been identified in LQTS. Hundreds of mutations have been described to cause these defects (see Table 2, right).8 Approximately 70% of congenital LQTS are caused by mutations in three genes and are classified as LQTS 1, LQTS 2, and LQTS 3.8 The other seven mutation types account for about 5% of cases; a quarter of LQTS cases have no identified genetic mutations.8
LQTS usually can be distinguished by clinical features and some ECG characteristics, but diagnosis of the specific type requires genetic testing.8,9 The most common types of LQTS are discussed below.
- Long QT1 is the most common type, occurring in approximately 40% to 50% of patients diagnosed with LQTS. It is characterized by a defect in the potassium channel alpha subunit leading to IKs reduction.9 These patients typically present with syncope or adrenergic-induced torsades, might have wide, broad-based T-waves on ECG, and respond well to beta-blocker therapy.6 Triggers for these patients include physical exertion or emotional stressors, particularly exercise and swimming. These patients typically present in early childhood.1
- Long QT2 occurs in 35% to 40% of patients and is characterized by a different defect in the alpha subunit of the potassium channel, which leads to reduced IKr.9 ECGs in LQTS2 can demonstrate low-amplitude and notched T-waves. Sudden catecholamine surges related to emotional stress or loud noises and bradycardia can trigger dysrhythmias in Long QT2.6 Thus, beta blockers reduce overall cardiac events in LQTS2 but less effectively than in LQTS1.6 These patients also present in childhood but typically are older than patients with LQTS1.6
- Long QT3 is less common than LQTS1 or LQTS2, at 2% to 8% of LQTS patients, but carries a higher mortality and is not treated effectively with beta blockers. LQTS3 is characterized by a defect in a sodium channel, causing a gain-of-function in the INa.4,9 These patients are more likely to have a fatal dysrhythmia while sleeping, are less susceptible to exercise-induced events, and have increased morbidity and mortality associated with bradycardia.4,9 ECG frequently reveals a relatively long ST segment, followed by a peaked and tall T-wave. Beta-blocker therapy can predispose to dysrhythmias in these patients; therefore, many of these patients will have pacemakers implanted as first-line therapy.6
While less common, Jervell and Lange Nielson syndrome is an autosomal recessive form of LQTS in which affected patients have homozygous mutations in the KCNQ1 or KCNE1 genes. This syndrome occurs in approximately 1% to 7% of LQTS patients, displays a typical QTc >550 ms, can be triggered by exercise and emotional stress, is associated with deafness, and carries a high risk of cardiac events at a young age.6
Acquired Syndromes
In addition to congenital LQTS, certain patients can acquire LQTS after being treated with particular drugs or having metabolic abnormalities, namely hypomagnesemia, hypocalcemia, and hypokalemia. Most experts think patients who “acquire” LQTS that predisposes to torsades de pointes have underlying structural heart disease or LQTS genetic carrier mutations that combine with transient initiating events (e.g., drugs or metabolic abnormalities) to induce dysrhythmias.1 In addition to certain drugs, cardiac ischemia, and electrolyte abnormalities, cocaine abuse, HIV, and subarachnoid hemorrhage can induce dysrhythmias in susceptible patients.5
Many types of drugs can cause a prolonged QT interval, and others should be avoided in patients with pre-existing prolonged QT (see Table 3, p. 17). Potentially offending drugs that are frequently encountered by inpatient physicians include amiodarone, diltiazem, erythromycin, clarithromycin, ciprofloxacin, fluoxetine, paroxetine, sertraline, haloperidol, ritonavir, and methadone.1 Additionally, drugs that cause electrolyte abnormalities (e.g., diuretics and lithium) should be monitored closely.
Overall, the goals of therapy in LQTS are:
- Decrease the risk of dysrhythmic events;
- Minimize adrenergic response;
- Shorten the QTc;
- Decrease the dispersion of refractoriness; and
- Improve the function of the ion channels.3
Supportive measures should be taken for patients who are acutely symptomatic from LQTS and associated torsades de pointes. In addition to immediate cardioversion for ongoing and hemodynamically significant torsades, intravenous magnesium should be administered, electrolytes corrected, and offending drugs discontinued.5 Temporary transvenous pacing at rates of approximately 100 beats per minute is highly effective in preventing short-term recurrence of torsades in congenital and acquired LQTS, especially in bradycardic patients.5 Isoproterenol infusion increases the heart rate and effectively prevents acute recurrence of torsades in patients with acquired LQTS, but it should be used with caution in patients with structural heart disease.5
Long-term strategies to manage LQTS include:
- Minimizing the risk of triggering cardiac events via adrenergic stimulation;
- Preventing ongoing dysrhythmias;
- Avoiding medications known to prolong the QT interval; and
- Maintaining normal electrolytes and minerals.5
Most patients with congenital long QT should be discouraged from participating in competitive sports, and patients should attempt to eliminate exposures to stress or sudden awakening, though this is not practical in all cases.5 Beta blockers generally are the first-line therapy and are more effective for LQT1 than LQT2 or LQT3.4,5 If patients are still symptomatic despite adequate medical therapy, or have survived cardiac arrest, they should be considered for ICD therapy.4,5 In addition, patients with profound bradycardia benefit from pacemaker implantation.5 Patients who remain symptomatic despite both beta blockade and ICD placement might find cervicothoracic sympathectomy curative.4,5
Perioperative Considerations
Although little data is available to guide physicians in the prevention of torsades de pointes during the course of anesthesia, there are a number of considerations that may reduce the chances of symptomatic dysrhythmias.
First, care should be taken to avoid dysrhythmia triggers in LQTS by providing a calm, quiet environment during induction, monitoring, and treating metabolic abnormalities, and providing an appropriate level of anesthesia.10 Beta-blocker therapy should be continued and potentially measured preoperatively by assessing heart rate response during stress testing.5 An implantable cardioverter-defibrillator (AICD) should be interrogated prior to surgery and inactivated during the operation.5
Finally, Kies et al have recommended general anesthesia with propofol for induction (or throughout), isoflurane as the volatile agent, vecuronium for muscle relaxation, and intravenous fentanyl for analgesia when possible.10
Back to the Case
While the patient had no genetic testing for LQTS, evaluation of previous ECGs demonstrated a prolonged QT interval. The hip fracture repair was considered an urgent procedure, which precluded the ability to undertake genetic testing and consideration for device implantation. The only medication that was known to increase the risk for dysrhythmias in this patient was his diuretic, with the attendant risk of electrolyte abnormalities.
Thus, the patient’s hydrochlorothiazide was discontinued and his pre-existing atenolol continued. The patient’s electrolytes and minerals were monitored closely, and magnesium was administered on the day of surgery. Anesthesia was made aware of the prolonged QT interval, such that they were able to minimize the risk for and anticipate the treatment of dysrhythmias. The patient tolerated the surgery and post-operative period without complication and was scheduled for an outpatient workup and management for his prolonged QT interval.
Bottom Line
Long QT syndrome is frequently genetic in origin, but it can be caused by certain medications and perturbations of electrolytes. Beta blockers are the first-line therapy for the majority of LQTS cases, along with discontinuation of drugs that might induce or aggravate the QT prolongation.
Patients who have had cardiac arrest or who remain symptomatic despite beta-blocker therapy should have an ICD implanted.
In the perioperative period, patients’ electrolytes should be monitored and kept within normal limits. If the patient is on a beta blocker, it should be continued, and the anesthesiologist should be made aware of the diagnosis so that the anesthethic plan can be optimized to prevent arrhythmic complications. TH
Dr. Kamali is a medical resident at the University of Colorado Denver. Dr. Stickrath is a hospitalist at the Veterans Affairs Medical Center in Denver and an instructor of medicine at UC Denver. Dr. Prochazka is director of ambulatory care at the Denver VA and professor of medicine at UC Denver. Dr. Varosy is director of cardiac electrophysiology at the Denver VA and assistant professor of medicine at UC Denver.
References
- Kao LW, Furbee BR. Drug-induced q-T prolongation. Med Clin North Am. 2005;89(6):1125-1144.
- Marchlinski F. Chapter 226, The Tachyarrhythmias; Harrison's Principles of Internal Medicine, 17e. Available at: www.accessmedicine.com/resourceTOC .aspx?resourceID=4. Accessed Nov. 21, 2009.
- Zareba W, Cygankiewicz I. Long QT syndrome and short QT syndrome. Prog Cardiovasc Dis. 2008; 51(3):264-278.
- Booker PD, Whyte SD, Ladusans EJ. Long QT syndrome and anaesthesia. Br J Anaesth. 2003;90(3):349-366.
- Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002;143(1):7-14.
- Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372(9640):750-763.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-784.
- Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752-1760.
- Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med. 2006;8(3):143-155.
- Kies SJ, Pabelick CM, Hurley HA, White RD, Ackerman MJ. Anesthesia for patients with congenital long QT syndrome. Anesthesiology. 2005;102(1):204-210.
- Wisely NA, Shipton EA. Long QT syndrome and anaesthesia. Eur J Anaesthesiol. 2002;19(12):853-859.
Case
You are asked to admit a 63-year-old male with a history of hypertension and osteoarthritis. The patient, who fell at home, is scheduled for open repair of his femoral neck fracture the following day. The patient reports tripping over his granddaughter’s toys and denies any associated symptoms around the time of his fall. An electrocardiogram (ECG) reveals a QTc (QT) interval of 480 ms. How should this hospitalized patient’s prolonged QT interval be managed?
Overview
Patients with a prolonged QT interval on routine ECG present an interesting dilemma for clinicians. Although QT prolongation—either congenital or acquired—has been associated with dysrhythmias, the risk of torsades de pointes and sudden cardiac death varies considerably based on myriad underlying factors.1 Therefore, the principle job of the clinician who has recognized QT prolongation is to assess and minimize the risk of the development of clinically significant dysrhythmias, and to be prepared to manage them should they arise.
The QT interval encompasses ventricular depolarization and repolarization. This ventricular action potential proceeds through five phases. The initial upstroke (phase 0) of depolarization occurs with the opening of Na+ channels, triggering the inward Na+ current (INa), and causes the interior of the myocytes to become positively charged. This is followed by initial repolarization (phase 1) when the opening of K+ channels causes an outward K+ current (Ito). Next, the plateau phase (phase 2) of the action potential follows with a balance of inward current through Ca2+channels (Ica-L) and outward current through slow rectifier K+ channels (IKs), and then later through delayed, rapid K+ rectifier channels (IKr). Then, the inward current is deactivated, while the outward current increases through the rapid delayed rectifier (IKr) and opening of inward rectifier channels (IK1) to complete repolarization (phase 3). Finally, the action potential returns to baseline (phase 4) and Na+ begins to enter the cell again (see Figure 1, above).
The long QT syndrome (LQTS) is defined by a defect in these cardiac ion channels, which leads to abnormal repolarization, usually lengthening the QT interval and thus predisposing to ventricular dysrhythmias.2 It is estimated that as many as 85% of these syndromes are inherited, and up to 15% are acquired or sporadic.3 Depending on the underlying etiology of the LQTS, manifestations might first be appreciated at any time from in utero through adulthood.4 Symptoms including palpitations, syncope, seizures, or cardiac arrest bring these patients to medical attention.3 These symptoms frequently elicit physical or emotional stress, but they can occur without obvious inciting triggers.5 A 20% mortality risk exists in patients who are symptomatic and untreated in the first year following diagnosis, and up to 50% within 10 years following diagnosis.4
How is Long QT Syndrome Diagnosed?
The LQTS diagnosis is based on clinical history in combination with ECG abnormalities.6 Important historical elements include symptoms of palpitations, syncope, seizures, or cardiac arrest.3 In addition, a family history of unexplained syncope or sudden death, especially at a young age, should raise LQTS suspicion.5
A variety of ECG findings can be witnessed in LQTS patients.4,5 Although the majority of patients have a QTc >440 ms, approximately one-third have a QTc ≤460 ms, and about 10% have normal QTc intervals.5 Other ECG abnormalities include notched, biphasic, or prolonged T-waves, and the presence of U-waves.4,5 Schwartz et al used these elements to publish criteria (see Table 1, right) that physicians can use to assess the probability that a patient has LQTS.7
Types of Long QT Syndromes
Because the risk of developing significant dysrhythmias with LQTS is dependent on both the actual QT interval, with risk for sudden cardiac death increased two to three times with QT >440 ms compared with QT <440 ms and the specific underlying genotype, it is important to have an understanding of congenital and acquired LQTS and the associated triggers for torsades de pointes.
Congenital LQTS
Congenital LQTS is caused by mutations in cardiac ion channel proteins, primarily sodium, and potassium channels.5,6 These defects either slow depolarization or lengthen repolarization, leading to heterogeneity of repolarization of the membrane.5 This, in turn, predisposes to ventricular dysrhythmias, including torsades de pointes and subsequent ventricular fibrillation and death.2 Currently, 12 genetic defects have been identified in LQTS. Hundreds of mutations have been described to cause these defects (see Table 2, right).8 Approximately 70% of congenital LQTS are caused by mutations in three genes and are classified as LQTS 1, LQTS 2, and LQTS 3.8 The other seven mutation types account for about 5% of cases; a quarter of LQTS cases have no identified genetic mutations.8
LQTS usually can be distinguished by clinical features and some ECG characteristics, but diagnosis of the specific type requires genetic testing.8,9 The most common types of LQTS are discussed below.
- Long QT1 is the most common type, occurring in approximately 40% to 50% of patients diagnosed with LQTS. It is characterized by a defect in the potassium channel alpha subunit leading to IKs reduction.9 These patients typically present with syncope or adrenergic-induced torsades, might have wide, broad-based T-waves on ECG, and respond well to beta-blocker therapy.6 Triggers for these patients include physical exertion or emotional stressors, particularly exercise and swimming. These patients typically present in early childhood.1
- Long QT2 occurs in 35% to 40% of patients and is characterized by a different defect in the alpha subunit of the potassium channel, which leads to reduced IKr.9 ECGs in LQTS2 can demonstrate low-amplitude and notched T-waves. Sudden catecholamine surges related to emotional stress or loud noises and bradycardia can trigger dysrhythmias in Long QT2.6 Thus, beta blockers reduce overall cardiac events in LQTS2 but less effectively than in LQTS1.6 These patients also present in childhood but typically are older than patients with LQTS1.6
- Long QT3 is less common than LQTS1 or LQTS2, at 2% to 8% of LQTS patients, but carries a higher mortality and is not treated effectively with beta blockers. LQTS3 is characterized by a defect in a sodium channel, causing a gain-of-function in the INa.4,9 These patients are more likely to have a fatal dysrhythmia while sleeping, are less susceptible to exercise-induced events, and have increased morbidity and mortality associated with bradycardia.4,9 ECG frequently reveals a relatively long ST segment, followed by a peaked and tall T-wave. Beta-blocker therapy can predispose to dysrhythmias in these patients; therefore, many of these patients will have pacemakers implanted as first-line therapy.6
While less common, Jervell and Lange Nielson syndrome is an autosomal recessive form of LQTS in which affected patients have homozygous mutations in the KCNQ1 or KCNE1 genes. This syndrome occurs in approximately 1% to 7% of LQTS patients, displays a typical QTc >550 ms, can be triggered by exercise and emotional stress, is associated with deafness, and carries a high risk of cardiac events at a young age.6
Acquired Syndromes
In addition to congenital LQTS, certain patients can acquire LQTS after being treated with particular drugs or having metabolic abnormalities, namely hypomagnesemia, hypocalcemia, and hypokalemia. Most experts think patients who “acquire” LQTS that predisposes to torsades de pointes have underlying structural heart disease or LQTS genetic carrier mutations that combine with transient initiating events (e.g., drugs or metabolic abnormalities) to induce dysrhythmias.1 In addition to certain drugs, cardiac ischemia, and electrolyte abnormalities, cocaine abuse, HIV, and subarachnoid hemorrhage can induce dysrhythmias in susceptible patients.5
Many types of drugs can cause a prolonged QT interval, and others should be avoided in patients with pre-existing prolonged QT (see Table 3, p. 17). Potentially offending drugs that are frequently encountered by inpatient physicians include amiodarone, diltiazem, erythromycin, clarithromycin, ciprofloxacin, fluoxetine, paroxetine, sertraline, haloperidol, ritonavir, and methadone.1 Additionally, drugs that cause electrolyte abnormalities (e.g., diuretics and lithium) should be monitored closely.
Overall, the goals of therapy in LQTS are:
- Decrease the risk of dysrhythmic events;
- Minimize adrenergic response;
- Shorten the QTc;
- Decrease the dispersion of refractoriness; and
- Improve the function of the ion channels.3
Supportive measures should be taken for patients who are acutely symptomatic from LQTS and associated torsades de pointes. In addition to immediate cardioversion for ongoing and hemodynamically significant torsades, intravenous magnesium should be administered, electrolytes corrected, and offending drugs discontinued.5 Temporary transvenous pacing at rates of approximately 100 beats per minute is highly effective in preventing short-term recurrence of torsades in congenital and acquired LQTS, especially in bradycardic patients.5 Isoproterenol infusion increases the heart rate and effectively prevents acute recurrence of torsades in patients with acquired LQTS, but it should be used with caution in patients with structural heart disease.5
Long-term strategies to manage LQTS include:
- Minimizing the risk of triggering cardiac events via adrenergic stimulation;
- Preventing ongoing dysrhythmias;
- Avoiding medications known to prolong the QT interval; and
- Maintaining normal electrolytes and minerals.5
Most patients with congenital long QT should be discouraged from participating in competitive sports, and patients should attempt to eliminate exposures to stress or sudden awakening, though this is not practical in all cases.5 Beta blockers generally are the first-line therapy and are more effective for LQT1 than LQT2 or LQT3.4,5 If patients are still symptomatic despite adequate medical therapy, or have survived cardiac arrest, they should be considered for ICD therapy.4,5 In addition, patients with profound bradycardia benefit from pacemaker implantation.5 Patients who remain symptomatic despite both beta blockade and ICD placement might find cervicothoracic sympathectomy curative.4,5
Perioperative Considerations
Although little data is available to guide physicians in the prevention of torsades de pointes during the course of anesthesia, there are a number of considerations that may reduce the chances of symptomatic dysrhythmias.
First, care should be taken to avoid dysrhythmia triggers in LQTS by providing a calm, quiet environment during induction, monitoring, and treating metabolic abnormalities, and providing an appropriate level of anesthesia.10 Beta-blocker therapy should be continued and potentially measured preoperatively by assessing heart rate response during stress testing.5 An implantable cardioverter-defibrillator (AICD) should be interrogated prior to surgery and inactivated during the operation.5
Finally, Kies et al have recommended general anesthesia with propofol for induction (or throughout), isoflurane as the volatile agent, vecuronium for muscle relaxation, and intravenous fentanyl for analgesia when possible.10
Back to the Case
While the patient had no genetic testing for LQTS, evaluation of previous ECGs demonstrated a prolonged QT interval. The hip fracture repair was considered an urgent procedure, which precluded the ability to undertake genetic testing and consideration for device implantation. The only medication that was known to increase the risk for dysrhythmias in this patient was his diuretic, with the attendant risk of electrolyte abnormalities.
Thus, the patient’s hydrochlorothiazide was discontinued and his pre-existing atenolol continued. The patient’s electrolytes and minerals were monitored closely, and magnesium was administered on the day of surgery. Anesthesia was made aware of the prolonged QT interval, such that they were able to minimize the risk for and anticipate the treatment of dysrhythmias. The patient tolerated the surgery and post-operative period without complication and was scheduled for an outpatient workup and management for his prolonged QT interval.
Bottom Line
Long QT syndrome is frequently genetic in origin, but it can be caused by certain medications and perturbations of electrolytes. Beta blockers are the first-line therapy for the majority of LQTS cases, along with discontinuation of drugs that might induce or aggravate the QT prolongation.
Patients who have had cardiac arrest or who remain symptomatic despite beta-blocker therapy should have an ICD implanted.
In the perioperative period, patients’ electrolytes should be monitored and kept within normal limits. If the patient is on a beta blocker, it should be continued, and the anesthesiologist should be made aware of the diagnosis so that the anesthethic plan can be optimized to prevent arrhythmic complications. TH
Dr. Kamali is a medical resident at the University of Colorado Denver. Dr. Stickrath is a hospitalist at the Veterans Affairs Medical Center in Denver and an instructor of medicine at UC Denver. Dr. Prochazka is director of ambulatory care at the Denver VA and professor of medicine at UC Denver. Dr. Varosy is director of cardiac electrophysiology at the Denver VA and assistant professor of medicine at UC Denver.
References
- Kao LW, Furbee BR. Drug-induced q-T prolongation. Med Clin North Am. 2005;89(6):1125-1144.
- Marchlinski F. Chapter 226, The Tachyarrhythmias; Harrison's Principles of Internal Medicine, 17e. Available at: www.accessmedicine.com/resourceTOC .aspx?resourceID=4. Accessed Nov. 21, 2009.
- Zareba W, Cygankiewicz I. Long QT syndrome and short QT syndrome. Prog Cardiovasc Dis. 2008; 51(3):264-278.
- Booker PD, Whyte SD, Ladusans EJ. Long QT syndrome and anaesthesia. Br J Anaesth. 2003;90(3):349-366.
- Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002;143(1):7-14.
- Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372(9640):750-763.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-784.
- Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752-1760.
- Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med. 2006;8(3):143-155.
- Kies SJ, Pabelick CM, Hurley HA, White RD, Ackerman MJ. Anesthesia for patients with congenital long QT syndrome. Anesthesiology. 2005;102(1):204-210.
- Wisely NA, Shipton EA. Long QT syndrome and anaesthesia. Eur J Anaesthesiol. 2002;19(12):853-859.