User login
Painful Retiform Purpura in a Peritoneal Dialysis Patient
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
.")
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
A 72-year-old woman presented to the emergency department with concerns of confusion and lethargy during a session of peritoneal dialysis, which she had been receiving for the last 2 years for end-stage renal disease. She had a history of type 2 diabetes mellitus, diabetic retinopathy, hypertension, coronary artery disease, and peripheral vascular disease preceding a recent right below-knee amputation. A review of systems was positive for a rash on the thighs of several weeks’ duration that was preceded by several days of burning pain in the same distribution. Physical examination revealed retiform purpura with irregular contours and interspersed white stellate patterns scattered across the superomedial thighs, right lower back, and left lower abdomen. An initial laboratory workup revealed an elevated creatinine level of 5.03 mg/dL (reference range, 0.6–1.1 mg/dL; baseline level, 3.0 mg/dL) and mild leukocytosis (12.5 cells/mm3 [reference range, 4.5–11.0 cells/mm3]). Dermatology was consulted, and a 4-mm punch biopsy was obtained from the left medial thigh. Nephrology, infectious disease, and wound care consultations also were placed.


Atypical Fibroxanthoma Arising Within Erosive Pustular Dermatosis of the Scalp
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
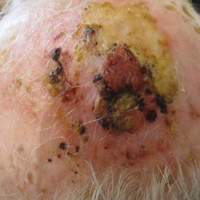
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
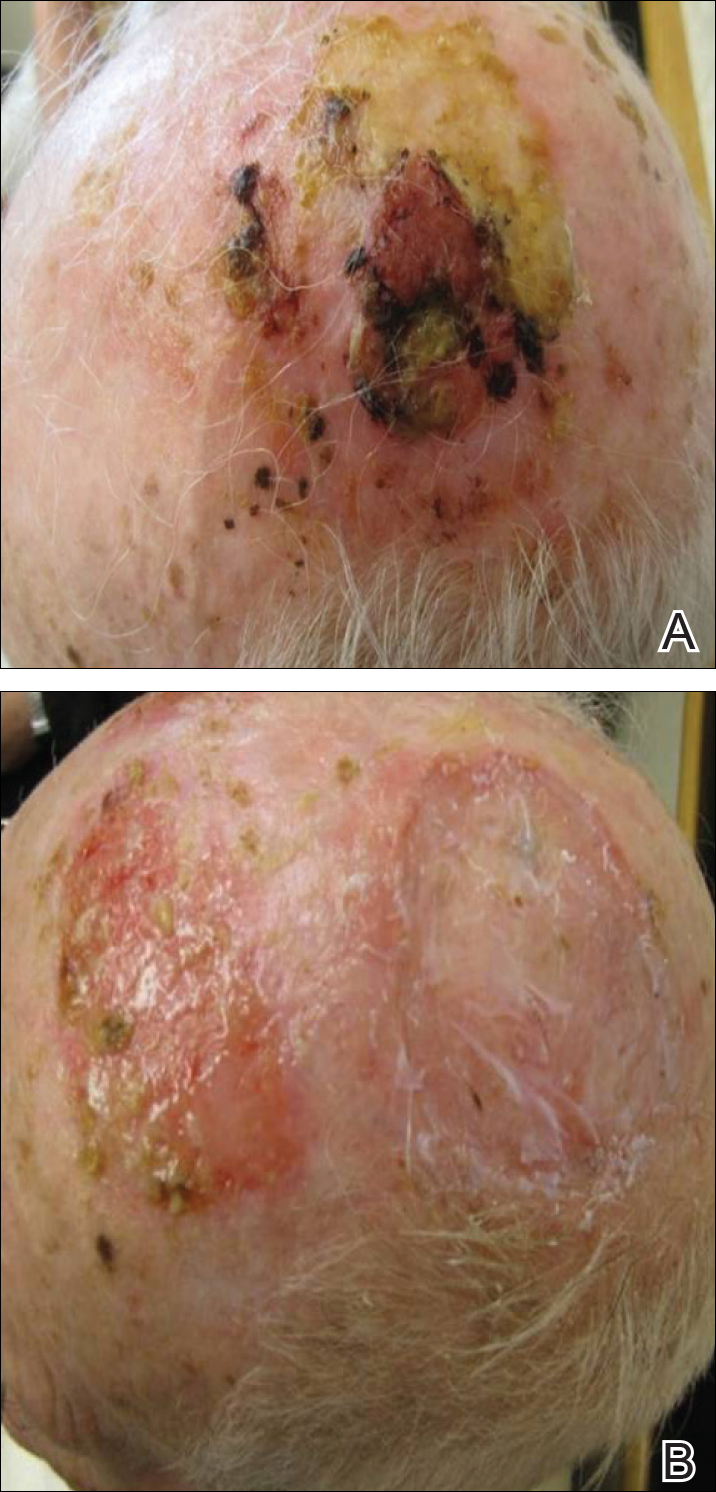
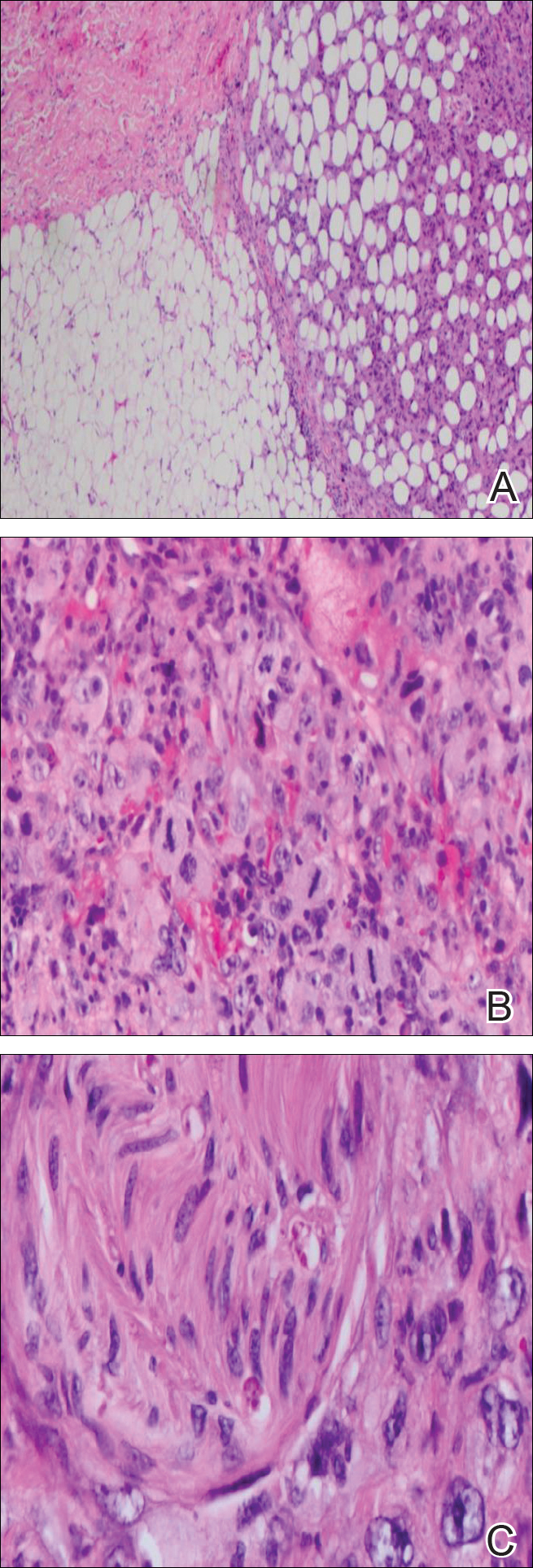
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
Practice Points
- Atypical fibroxanthoma predominantly occurs in older men on the head and neck.
- Erosive pustular dermatosis may be a benign entity, but if it does not resolve, continue to rebiopsy, as rare tumors may mimic this condition.