User login
Clinical Conundrum
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
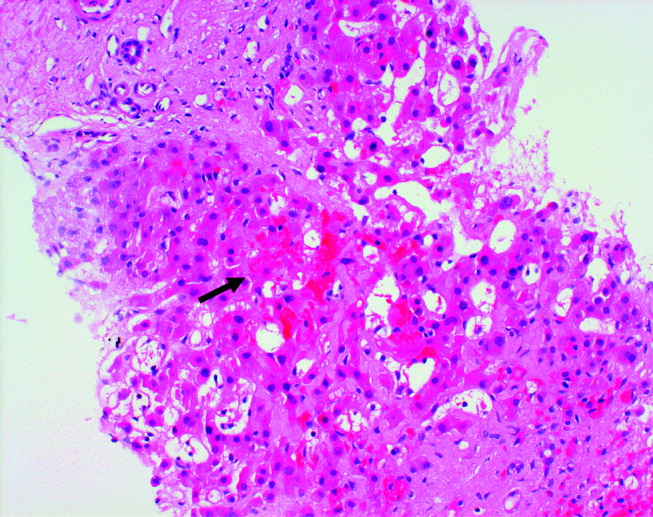
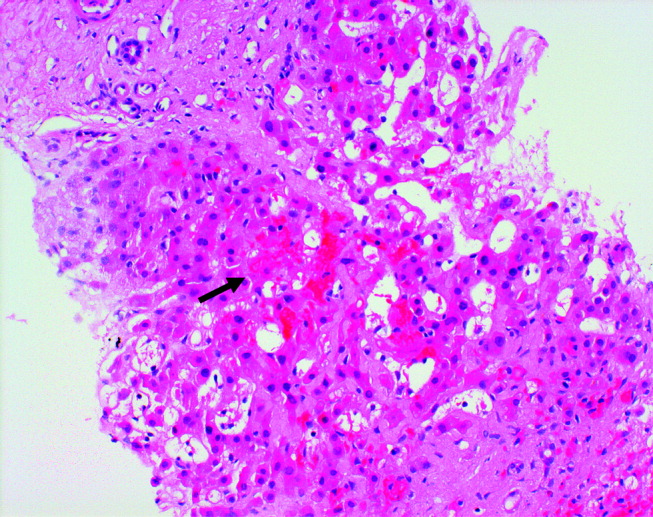
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
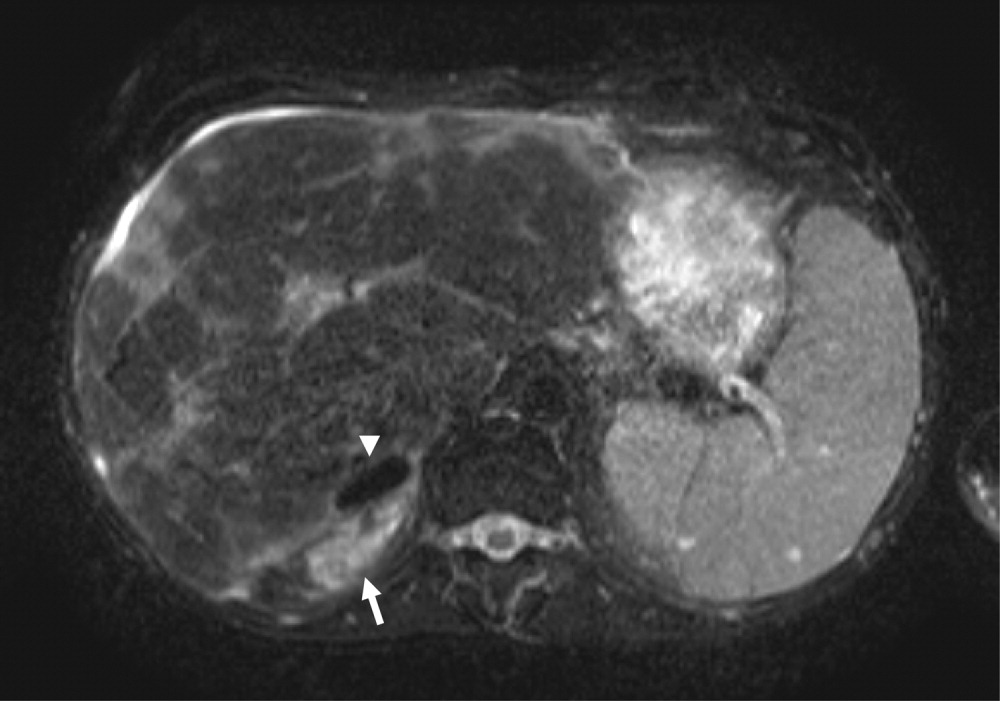
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.