User login
Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
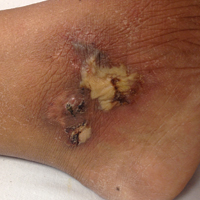
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.
Antiphospholipid Syndrome in a Patient With Rheumatoid Arthritis
Case Report
A 39-year-old woman with a 20-year history of rheumatoid arthritis (RA) presented to a university-affiliated tertiary care hospital with painful ulcerations on the bilateral dorsal feet that started as bullae 16 weeks prior to presentation. Initial skin biopsy performed by an outside dermatologist 8 weeks prior to presentation showed vasculitis and culture was positive for methicillin-sensitive Staphylococcus aureus. She was started on a prednisone taper and cephalexin, which did not improve the lower extremity ulcerations and the pain became progressively worse. At the time of presentation to our dermatology department, the patient was taking prednisone, hydroxychloroquine, hydrocodone-acetaminophen, and gabapentin. Prior therapy with sulfasalazine failed; etanercept and methotrexate were discontinued years prior due to side effects. The patient had no history of deep vein thrombosis, pulmonary embolism, or miscarriage.
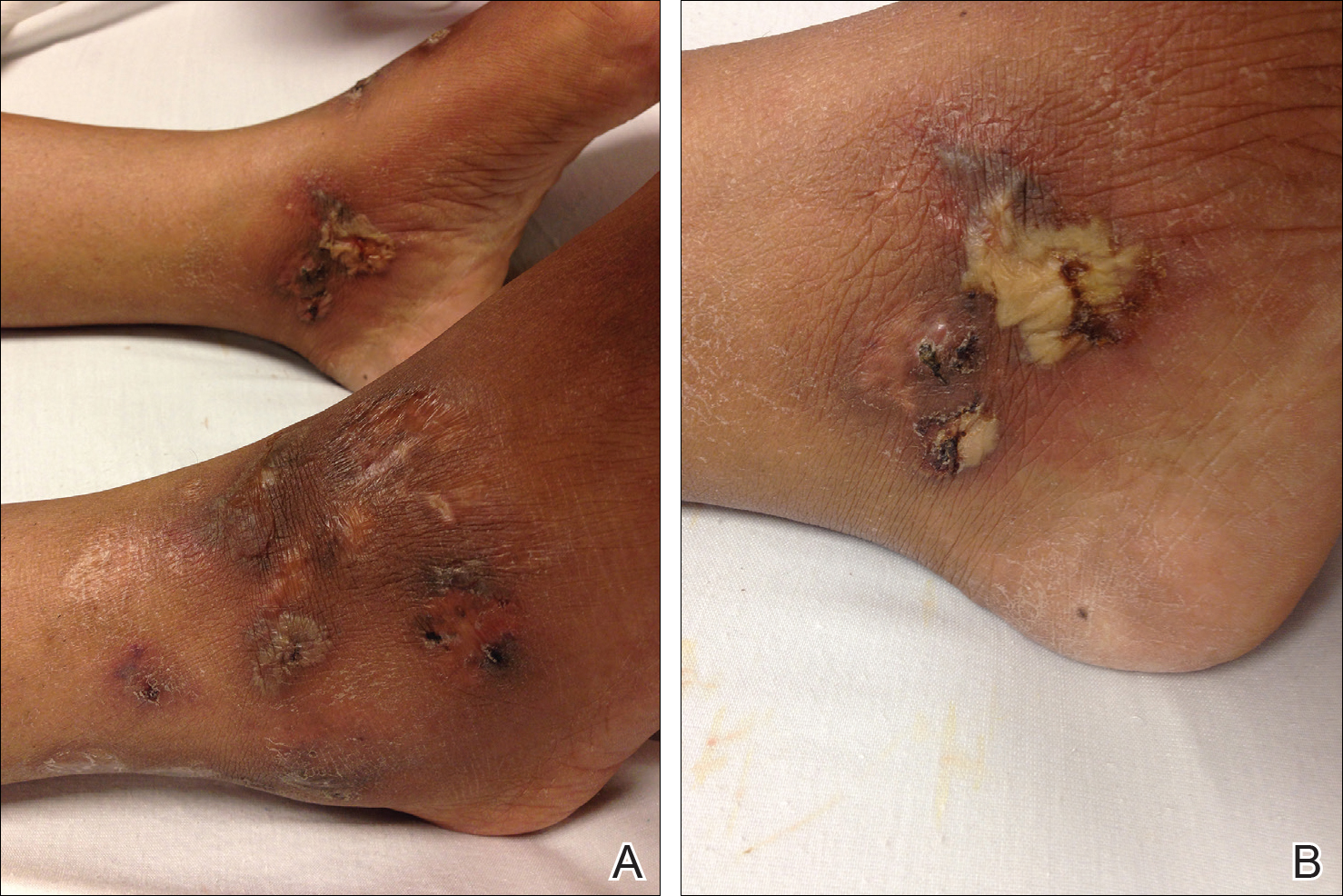
At presentation, the patient was afebrile and her vital signs were stable. Physical examination showed multiple ulcers and erosions on the bilateral dorsal feet with a few scattered retiform red-purple patches (Figure). One bulla was present on the right dorsal foot. All lesions were tender to the touch and edema was present on the bilateral feet. No oral ulcerations were present and no focal neuropathies or palpable cords were appreciated in the lower extremities. There were no other cutaneous abnormalities.
Laboratory studies showed a white blood cell count of 9.54×103/µL (reference range, 4.16-9.95×103/µL), hemoglobin count of 12.4 g/dL (reference range, 11.6-15.2 g/dL), and a platelet count of 175×103/µL (reference range, 143-398×103/µL). A basic metabolic panel was normal except for an elevated glucose level of 185 mg/dL (reference range, 65-100 mg/dL). Urinalysis was normal. Erythrocyte sedimentation rate and C-reactive protein level were not elevated. Antinuclear antibodies and double-stranded DNA antibodies were normal. Prothrombin time was 10.4 seconds (reference range, 9.2-11.5 seconds) and dilute viper's venom time was negative. Rheumatoid factor level was elevated at 76 IU/mL (reference range, <25 IU/mL) and anti-citrullinated peptide antibody was moderately elevated at 42 U/mL (negative, <20 U/mL; weak positive, 20-39 U/mL; moderate positive, 40-59 U/mL; strong positive, >59 U/mL). The cardiolipin antibodies IgG, IgM, and IgA were within reference range. Results of β2-glycoprotein I IgG and IgM antibody tests were normal, but IgA was elevated at 34 µg/mL (reference range, <20 µg/mL). Wound cultures grew moderate Enterobacter cloacae and Staphylococcus lugdunensis.
Slides from 2 prior punch biopsies obtained by an outside hospital approximately 8 weeks prior from the right and left dorsal foot lesions were reviewed. Both biopsies were histologically similar. Postcapillary venules showed extensive vasculitis with numerous fibrin thrombi in the lumens in both biopsy specimens. The biopsy from the right foot showed prominent ulceration of the epidermis, with a few of the affected vessels showing minimal accompanying nuclear dust; however, the predominant pattern was not that of leukocytoclastic vasculitis. Biopsy from the left foot showed prominent epidermal necrosis with focal reepithelialization and scattered eosinophils. The pathologist felt that a vasculitis secondary to coagulopathy was most likely but that a drug reaction and rheumatoid vasculitis would be other entities to consider in the differential. A review of the laboratory findings from the outside hospital from approximately 12 weeks prior to presentation showed IgM was normal but IgG was elevated at 28 U/mL (reference range, 0-15 U/mL) and IgA was elevated at 8 U/mL (reference range, 0-7 U/mL); β2-glycoprotein I IgG antibodies were elevated at 37 mg/dL (reference range, 0-25.0 mg/dL) and β2-glycoprotein I IgA antibodies were elevated at 5 mg/dL (reference range, 0-4.0 mg/dL).
The clinical suspicion of a thrombotic event on the dorsal feet, which was confirmed histologically, and the persistently positive antiphospholipid (aPL) antibody titers helped to establish the diagnosis of antiphospholipid syndrome (APS) in the setting of RA. The dose of prednisone was increased from 10 mg daily on admission to 40 mg daily. The patient was started on enoxaparin 60 mg subcutaneously twice daily at initial presentation and was bridged to oral warfarin 2 mg daily after the diagnosis of APS was established. Oral doxycycline 100 mg twice daily was started for wound infection. The ulcerations gradually improved over the course of her 7-day hospitalization. She was continued on prednisone, hydroxychloroquine, and warfarin as an outpatient and has had no recurrence of lesions after 3 years of follow-up on this regimen.
Comment
Antiphospholipid syndrome is an autoimmune condition defined by a venous and/or arterial thrombotic event and/or pregnancy morbidity in the presence of persistently elevated aPL antibody titers. The most frequently detected subgroups of aPL are anticardiolipin (aCL) antibodies, anti-β2-glycoprotein I antibodies, and lupus anticoagulants.1 Primary APS occurs as an isolated entity, whereas secondary APS occurs in the setting of a preexisting autoimmune disease, infection, malignancy, or medication.2 The diagnostic criteria for APS requires positive aPL titers at least 12 weeks apart and a clinically confirmed thrombotic event or pregnancy morbidity.3
About one-third to half of patients with APS exhibit cutaneous manifestations.4,5 Livedo reticularis is most commonly observed and represents the first clinical sign of APS in 17.5% of cases.6 Cutaneous findings of APS also include anetoderma, cutaneous ulceration and necrosis, necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, purpura, ecchymoses, painful skin nodules, and subungual hemorrhages.7 The various cutaneous manifestations of APS are associated with a range of histopathologic findings, but noninflammatory thrombosis in small arteries and/or veins in the dermis and subcutaneous fat tissue is the most common histologic feature.4 Our patient exhibited cutaneous ulceration and necrosis, and biopsy clearly showed the presence of vasculitis and fibrin thrombi within postcapillary venules. These findings along with the persistently elevated β2-glycoprotein I IgA solidified the diagnosis of APS.
The most common cutaneous manifestations of RA are nodules (32%), Raynaud phenomenon (10%), and vasculitis (3%).8 The mean prevalence of aPL antibodies in patients with RA is 28%, though reports range from 5% to 75%.1 The presence of aPL or aCL does not predict the development of thrombosis and/or thrombocytopenia in RA patients9,10; however, aCL antibodies in RA patients are associated with a higher risk for developing rheumatoid nodules. It is hypothesized that the majority of aCL antibodies identified in RA patients have different specificities than those identified in other diseases that are associated with thrombotic events.1
Anticoagulation has been proven to decrease the risk for recurrent thrombotic events in patients with APS.11 Patients should discontinue the use of estrogen-containing oral contraceptives; avoid smoking cigarettes; and treat hypertension, hyperlipidemia, and diabetes mellitus, if present. The type and duration of anticoagulation therapy, especially for the treatment of the cutaneous manifestations of APS, is less well defined. Antiplatelet therapies such as low-dose aspirin or dipyridamole often are used for less severe cutaneous manifestations such as livedoid vasculopathy. Warfarin with a target international normalized ratio of 2.0 to 3.0 is most commonly used following major thrombotic events, including cutaneous necrosis and digital gangrene. The role of corticosteroids and immunosuppressants is unclear; one study showed that these therapies did not prevent further thrombotic events in patients with systemic lupus erythematosus.4
Conclusion
Although aPL antibodies are most prevalent in patients with systemic lupus erythematosus, an estimated 28% of patients with RA have elevated aPL titers. The aPL antibodies recognized in RA patients are thought to have a different specificity than those recognized in other APS-associated diseases because elevated aPL antibody titers are not associated with an increased incidence of thrombotic events in RA patients; however, larger studies are needed to clarify this phenomenon. It remains to be determined if this case of APS and RA represents a coincidence or a true disease association, but the recognition of the cutaneous and histological features of APS is crucial for establishing a diagnosis and initiating anticoagulation therapy to prevent further morbidity and mortality.
- Olech E, Merrill JT. The prevalence and clinical significance of antiphospholipid antibodies in rheumatoid arthritis. Curr Rheumatol Rep. 2006;8:100-108.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Asherson A, Francès C, Iaccarino FL, et al. Theantiphospholipid antibody syndrome: diagnosis, skin manifestations and current therapy. Clin Exp Rheumatol. 2006;24(1 suppl 40):S46-S51.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019-1027.
- Francès C, Niang S, Laffitte E, et al. Dermatologic manifestations of antiphospholipid syndrome. two hundred consecutive cases. Arthritis Rheum. 2005;52:1785-1793.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6, pt 1):970-982.
- Young A. Extra-articular manifestations and complications of rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21:907-927.
- Palomo I, Pinochet C, Alarcón M, et al. Prevalence of antiphospholipid antibodies in Chilean patients with rheumatoid arthritis. J Clin Lab Anal. 2006;20:190-194.
- Wolf P, Gretler J, Aglas F, et al. Anticardiolipin antibodies in rheumatoid arthritis: their relation to rheumatoid nodules and cutaneous vascular manifestations. Br J Dermatol. 1994;131:48-51.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA. 2006;295:1050-1057.
Case Report
A 39-year-old woman with a 20-year history of rheumatoid arthritis (RA) presented to a university-affiliated tertiary care hospital with painful ulcerations on the bilateral dorsal feet that started as bullae 16 weeks prior to presentation. Initial skin biopsy performed by an outside dermatologist 8 weeks prior to presentation showed vasculitis and culture was positive for methicillin-sensitive Staphylococcus aureus. She was started on a prednisone taper and cephalexin, which did not improve the lower extremity ulcerations and the pain became progressively worse. At the time of presentation to our dermatology department, the patient was taking prednisone, hydroxychloroquine, hydrocodone-acetaminophen, and gabapentin. Prior therapy with sulfasalazine failed; etanercept and methotrexate were discontinued years prior due to side effects. The patient had no history of deep vein thrombosis, pulmonary embolism, or miscarriage.
At presentation, the patient was afebrile and her vital signs were stable. Physical examination showed multiple ulcers and erosions on the bilateral dorsal feet with a few scattered retiform red-purple patches (Figure). One bulla was present on the right dorsal foot. All lesions were tender to the touch and edema was present on the bilateral feet. No oral ulcerations were present and no focal neuropathies or palpable cords were appreciated in the lower extremities. There were no other cutaneous abnormalities.
Laboratory studies showed a white blood cell count of 9.54×103/µL (reference range, 4.16-9.95×103/µL), hemoglobin count of 12.4 g/dL (reference range, 11.6-15.2 g/dL), and a platelet count of 175×103/µL (reference range, 143-398×103/µL). A basic metabolic panel was normal except for an elevated glucose level of 185 mg/dL (reference range, 65-100 mg/dL). Urinalysis was normal. Erythrocyte sedimentation rate and C-reactive protein level were not elevated. Antinuclear antibodies and double-stranded DNA antibodies were normal. Prothrombin time was 10.4 seconds (reference range, 9.2-11.5 seconds) and dilute viper's venom time was negative. Rheumatoid factor level was elevated at 76 IU/mL (reference range, <25 IU/mL) and anti-citrullinated peptide antibody was moderately elevated at 42 U/mL (negative, <20 U/mL; weak positive, 20-39 U/mL; moderate positive, 40-59 U/mL; strong positive, >59 U/mL). The cardiolipin antibodies IgG, IgM, and IgA were within reference range. Results of β2-glycoprotein I IgG and IgM antibody tests were normal, but IgA was elevated at 34 µg/mL (reference range, <20 µg/mL). Wound cultures grew moderate Enterobacter cloacae and Staphylococcus lugdunensis.
Slides from 2 prior punch biopsies obtained by an outside hospital approximately 8 weeks prior from the right and left dorsal foot lesions were reviewed. Both biopsies were histologically similar. Postcapillary venules showed extensive vasculitis with numerous fibrin thrombi in the lumens in both biopsy specimens. The biopsy from the right foot showed prominent ulceration of the epidermis, with a few of the affected vessels showing minimal accompanying nuclear dust; however, the predominant pattern was not that of leukocytoclastic vasculitis. Biopsy from the left foot showed prominent epidermal necrosis with focal reepithelialization and scattered eosinophils. The pathologist felt that a vasculitis secondary to coagulopathy was most likely but that a drug reaction and rheumatoid vasculitis would be other entities to consider in the differential. A review of the laboratory findings from the outside hospital from approximately 12 weeks prior to presentation showed IgM was normal but IgG was elevated at 28 U/mL (reference range, 0-15 U/mL) and IgA was elevated at 8 U/mL (reference range, 0-7 U/mL); β2-glycoprotein I IgG antibodies were elevated at 37 mg/dL (reference range, 0-25.0 mg/dL) and β2-glycoprotein I IgA antibodies were elevated at 5 mg/dL (reference range, 0-4.0 mg/dL).
The clinical suspicion of a thrombotic event on the dorsal feet, which was confirmed histologically, and the persistently positive antiphospholipid (aPL) antibody titers helped to establish the diagnosis of antiphospholipid syndrome (APS) in the setting of RA. The dose of prednisone was increased from 10 mg daily on admission to 40 mg daily. The patient was started on enoxaparin 60 mg subcutaneously twice daily at initial presentation and was bridged to oral warfarin 2 mg daily after the diagnosis of APS was established. Oral doxycycline 100 mg twice daily was started for wound infection. The ulcerations gradually improved over the course of her 7-day hospitalization. She was continued on prednisone, hydroxychloroquine, and warfarin as an outpatient and has had no recurrence of lesions after 3 years of follow-up on this regimen.
Comment
Antiphospholipid syndrome is an autoimmune condition defined by a venous and/or arterial thrombotic event and/or pregnancy morbidity in the presence of persistently elevated aPL antibody titers. The most frequently detected subgroups of aPL are anticardiolipin (aCL) antibodies, anti-β2-glycoprotein I antibodies, and lupus anticoagulants.1 Primary APS occurs as an isolated entity, whereas secondary APS occurs in the setting of a preexisting autoimmune disease, infection, malignancy, or medication.2 The diagnostic criteria for APS requires positive aPL titers at least 12 weeks apart and a clinically confirmed thrombotic event or pregnancy morbidity.3
About one-third to half of patients with APS exhibit cutaneous manifestations.4,5 Livedo reticularis is most commonly observed and represents the first clinical sign of APS in 17.5% of cases.6 Cutaneous findings of APS also include anetoderma, cutaneous ulceration and necrosis, necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, purpura, ecchymoses, painful skin nodules, and subungual hemorrhages.7 The various cutaneous manifestations of APS are associated with a range of histopathologic findings, but noninflammatory thrombosis in small arteries and/or veins in the dermis and subcutaneous fat tissue is the most common histologic feature.4 Our patient exhibited cutaneous ulceration and necrosis, and biopsy clearly showed the presence of vasculitis and fibrin thrombi within postcapillary venules. These findings along with the persistently elevated β2-glycoprotein I IgA solidified the diagnosis of APS.
The most common cutaneous manifestations of RA are nodules (32%), Raynaud phenomenon (10%), and vasculitis (3%).8 The mean prevalence of aPL antibodies in patients with RA is 28%, though reports range from 5% to 75%.1 The presence of aPL or aCL does not predict the development of thrombosis and/or thrombocytopenia in RA patients9,10; however, aCL antibodies in RA patients are associated with a higher risk for developing rheumatoid nodules. It is hypothesized that the majority of aCL antibodies identified in RA patients have different specificities than those identified in other diseases that are associated with thrombotic events.1
Anticoagulation has been proven to decrease the risk for recurrent thrombotic events in patients with APS.11 Patients should discontinue the use of estrogen-containing oral contraceptives; avoid smoking cigarettes; and treat hypertension, hyperlipidemia, and diabetes mellitus, if present. The type and duration of anticoagulation therapy, especially for the treatment of the cutaneous manifestations of APS, is less well defined. Antiplatelet therapies such as low-dose aspirin or dipyridamole often are used for less severe cutaneous manifestations such as livedoid vasculopathy. Warfarin with a target international normalized ratio of 2.0 to 3.0 is most commonly used following major thrombotic events, including cutaneous necrosis and digital gangrene. The role of corticosteroids and immunosuppressants is unclear; one study showed that these therapies did not prevent further thrombotic events in patients with systemic lupus erythematosus.4
Conclusion
Although aPL antibodies are most prevalent in patients with systemic lupus erythematosus, an estimated 28% of patients with RA have elevated aPL titers. The aPL antibodies recognized in RA patients are thought to have a different specificity than those recognized in other APS-associated diseases because elevated aPL antibody titers are not associated with an increased incidence of thrombotic events in RA patients; however, larger studies are needed to clarify this phenomenon. It remains to be determined if this case of APS and RA represents a coincidence or a true disease association, but the recognition of the cutaneous and histological features of APS is crucial for establishing a diagnosis and initiating anticoagulation therapy to prevent further morbidity and mortality.
Case Report
A 39-year-old woman with a 20-year history of rheumatoid arthritis (RA) presented to a university-affiliated tertiary care hospital with painful ulcerations on the bilateral dorsal feet that started as bullae 16 weeks prior to presentation. Initial skin biopsy performed by an outside dermatologist 8 weeks prior to presentation showed vasculitis and culture was positive for methicillin-sensitive Staphylococcus aureus. She was started on a prednisone taper and cephalexin, which did not improve the lower extremity ulcerations and the pain became progressively worse. At the time of presentation to our dermatology department, the patient was taking prednisone, hydroxychloroquine, hydrocodone-acetaminophen, and gabapentin. Prior therapy with sulfasalazine failed; etanercept and methotrexate were discontinued years prior due to side effects. The patient had no history of deep vein thrombosis, pulmonary embolism, or miscarriage.
At presentation, the patient was afebrile and her vital signs were stable. Physical examination showed multiple ulcers and erosions on the bilateral dorsal feet with a few scattered retiform red-purple patches (Figure). One bulla was present on the right dorsal foot. All lesions were tender to the touch and edema was present on the bilateral feet. No oral ulcerations were present and no focal neuropathies or palpable cords were appreciated in the lower extremities. There were no other cutaneous abnormalities.
Laboratory studies showed a white blood cell count of 9.54×103/µL (reference range, 4.16-9.95×103/µL), hemoglobin count of 12.4 g/dL (reference range, 11.6-15.2 g/dL), and a platelet count of 175×103/µL (reference range, 143-398×103/µL). A basic metabolic panel was normal except for an elevated glucose level of 185 mg/dL (reference range, 65-100 mg/dL). Urinalysis was normal. Erythrocyte sedimentation rate and C-reactive protein level were not elevated. Antinuclear antibodies and double-stranded DNA antibodies were normal. Prothrombin time was 10.4 seconds (reference range, 9.2-11.5 seconds) and dilute viper's venom time was negative. Rheumatoid factor level was elevated at 76 IU/mL (reference range, <25 IU/mL) and anti-citrullinated peptide antibody was moderately elevated at 42 U/mL (negative, <20 U/mL; weak positive, 20-39 U/mL; moderate positive, 40-59 U/mL; strong positive, >59 U/mL). The cardiolipin antibodies IgG, IgM, and IgA were within reference range. Results of β2-glycoprotein I IgG and IgM antibody tests were normal, but IgA was elevated at 34 µg/mL (reference range, <20 µg/mL). Wound cultures grew moderate Enterobacter cloacae and Staphylococcus lugdunensis.
Slides from 2 prior punch biopsies obtained by an outside hospital approximately 8 weeks prior from the right and left dorsal foot lesions were reviewed. Both biopsies were histologically similar. Postcapillary venules showed extensive vasculitis with numerous fibrin thrombi in the lumens in both biopsy specimens. The biopsy from the right foot showed prominent ulceration of the epidermis, with a few of the affected vessels showing minimal accompanying nuclear dust; however, the predominant pattern was not that of leukocytoclastic vasculitis. Biopsy from the left foot showed prominent epidermal necrosis with focal reepithelialization and scattered eosinophils. The pathologist felt that a vasculitis secondary to coagulopathy was most likely but that a drug reaction and rheumatoid vasculitis would be other entities to consider in the differential. A review of the laboratory findings from the outside hospital from approximately 12 weeks prior to presentation showed IgM was normal but IgG was elevated at 28 U/mL (reference range, 0-15 U/mL) and IgA was elevated at 8 U/mL (reference range, 0-7 U/mL); β2-glycoprotein I IgG antibodies were elevated at 37 mg/dL (reference range, 0-25.0 mg/dL) and β2-glycoprotein I IgA antibodies were elevated at 5 mg/dL (reference range, 0-4.0 mg/dL).
The clinical suspicion of a thrombotic event on the dorsal feet, which was confirmed histologically, and the persistently positive antiphospholipid (aPL) antibody titers helped to establish the diagnosis of antiphospholipid syndrome (APS) in the setting of RA. The dose of prednisone was increased from 10 mg daily on admission to 40 mg daily. The patient was started on enoxaparin 60 mg subcutaneously twice daily at initial presentation and was bridged to oral warfarin 2 mg daily after the diagnosis of APS was established. Oral doxycycline 100 mg twice daily was started for wound infection. The ulcerations gradually improved over the course of her 7-day hospitalization. She was continued on prednisone, hydroxychloroquine, and warfarin as an outpatient and has had no recurrence of lesions after 3 years of follow-up on this regimen.
Comment
Antiphospholipid syndrome is an autoimmune condition defined by a venous and/or arterial thrombotic event and/or pregnancy morbidity in the presence of persistently elevated aPL antibody titers. The most frequently detected subgroups of aPL are anticardiolipin (aCL) antibodies, anti-β2-glycoprotein I antibodies, and lupus anticoagulants.1 Primary APS occurs as an isolated entity, whereas secondary APS occurs in the setting of a preexisting autoimmune disease, infection, malignancy, or medication.2 The diagnostic criteria for APS requires positive aPL titers at least 12 weeks apart and a clinically confirmed thrombotic event or pregnancy morbidity.3
About one-third to half of patients with APS exhibit cutaneous manifestations.4,5 Livedo reticularis is most commonly observed and represents the first clinical sign of APS in 17.5% of cases.6 Cutaneous findings of APS also include anetoderma, cutaneous ulceration and necrosis, necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, purpura, ecchymoses, painful skin nodules, and subungual hemorrhages.7 The various cutaneous manifestations of APS are associated with a range of histopathologic findings, but noninflammatory thrombosis in small arteries and/or veins in the dermis and subcutaneous fat tissue is the most common histologic feature.4 Our patient exhibited cutaneous ulceration and necrosis, and biopsy clearly showed the presence of vasculitis and fibrin thrombi within postcapillary venules. These findings along with the persistently elevated β2-glycoprotein I IgA solidified the diagnosis of APS.
The most common cutaneous manifestations of RA are nodules (32%), Raynaud phenomenon (10%), and vasculitis (3%).8 The mean prevalence of aPL antibodies in patients with RA is 28%, though reports range from 5% to 75%.1 The presence of aPL or aCL does not predict the development of thrombosis and/or thrombocytopenia in RA patients9,10; however, aCL antibodies in RA patients are associated with a higher risk for developing rheumatoid nodules. It is hypothesized that the majority of aCL antibodies identified in RA patients have different specificities than those identified in other diseases that are associated with thrombotic events.1
Anticoagulation has been proven to decrease the risk for recurrent thrombotic events in patients with APS.11 Patients should discontinue the use of estrogen-containing oral contraceptives; avoid smoking cigarettes; and treat hypertension, hyperlipidemia, and diabetes mellitus, if present. The type and duration of anticoagulation therapy, especially for the treatment of the cutaneous manifestations of APS, is less well defined. Antiplatelet therapies such as low-dose aspirin or dipyridamole often are used for less severe cutaneous manifestations such as livedoid vasculopathy. Warfarin with a target international normalized ratio of 2.0 to 3.0 is most commonly used following major thrombotic events, including cutaneous necrosis and digital gangrene. The role of corticosteroids and immunosuppressants is unclear; one study showed that these therapies did not prevent further thrombotic events in patients with systemic lupus erythematosus.4
Conclusion
Although aPL antibodies are most prevalent in patients with systemic lupus erythematosus, an estimated 28% of patients with RA have elevated aPL titers. The aPL antibodies recognized in RA patients are thought to have a different specificity than those recognized in other APS-associated diseases because elevated aPL antibody titers are not associated with an increased incidence of thrombotic events in RA patients; however, larger studies are needed to clarify this phenomenon. It remains to be determined if this case of APS and RA represents a coincidence or a true disease association, but the recognition of the cutaneous and histological features of APS is crucial for establishing a diagnosis and initiating anticoagulation therapy to prevent further morbidity and mortality.
- Olech E, Merrill JT. The prevalence and clinical significance of antiphospholipid antibodies in rheumatoid arthritis. Curr Rheumatol Rep. 2006;8:100-108.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Asherson A, Francès C, Iaccarino FL, et al. Theantiphospholipid antibody syndrome: diagnosis, skin manifestations and current therapy. Clin Exp Rheumatol. 2006;24(1 suppl 40):S46-S51.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019-1027.
- Francès C, Niang S, Laffitte E, et al. Dermatologic manifestations of antiphospholipid syndrome. two hundred consecutive cases. Arthritis Rheum. 2005;52:1785-1793.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6, pt 1):970-982.
- Young A. Extra-articular manifestations and complications of rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21:907-927.
- Palomo I, Pinochet C, Alarcón M, et al. Prevalence of antiphospholipid antibodies in Chilean patients with rheumatoid arthritis. J Clin Lab Anal. 2006;20:190-194.
- Wolf P, Gretler J, Aglas F, et al. Anticardiolipin antibodies in rheumatoid arthritis: their relation to rheumatoid nodules and cutaneous vascular manifestations. Br J Dermatol. 1994;131:48-51.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA. 2006;295:1050-1057.
- Olech E, Merrill JT. The prevalence and clinical significance of antiphospholipid antibodies in rheumatoid arthritis. Curr Rheumatol Rep. 2006;8:100-108.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Asherson A, Francès C, Iaccarino FL, et al. Theantiphospholipid antibody syndrome: diagnosis, skin manifestations and current therapy. Clin Exp Rheumatol. 2006;24(1 suppl 40):S46-S51.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019-1027.
- Francès C, Niang S, Laffitte E, et al. Dermatologic manifestations of antiphospholipid syndrome. two hundred consecutive cases. Arthritis Rheum. 2005;52:1785-1793.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6, pt 1):970-982.
- Young A. Extra-articular manifestations and complications of rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21:907-927.
- Palomo I, Pinochet C, Alarcón M, et al. Prevalence of antiphospholipid antibodies in Chilean patients with rheumatoid arthritis. J Clin Lab Anal. 2006;20:190-194.
- Wolf P, Gretler J, Aglas F, et al. Anticardiolipin antibodies in rheumatoid arthritis: their relation to rheumatoid nodules and cutaneous vascular manifestations. Br J Dermatol. 1994;131:48-51.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA. 2006;295:1050-1057.
Practice Points
- Antiphospholipid syndrome (APS) is an autoimmune condition defined by a venous and/or arterial thrombotic event and/or pregnancy morbidity in the presence of persistently elevated antiphospholipid antibody titers.
- Cutaneous findings of APS include livedo reticularis most commonly but also anetoderma, cutaneous ulceration and necrosis, necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, purpura, ecchymoses, painful skin nodules, and subungual hemorrhages.
- The various cutaneous manifestations of APS are associated with a range of histopathologic findings, but noninflammatory thrombosis in small arteries and/or veins in the dermis and subcutaneous fat tissue is the most common histologic feature.