Article
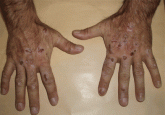
Lesions on the hands, high aminotransferase levels
A 64-year-old man presents with a 10-day history of painful vesicles and erosions on the dorsa of the hands that appeared after sun exposure. What...
Article

Painful eye with a facial rash
Six days ago, the patient had an intense headache; 4 days ago, painful herpetiform lesions appeared on his face. What is the diagnosis?
Article
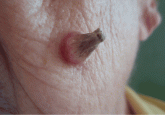
A facial cutaneous horn
A healthy 84-year-old woman has an asymptomatic lesion on her cheek that has grown progressively over the past 10 months. What is it?
Article
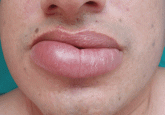
A persistently swollen lip
An otherwise healthy man has had asymptomatic swelling of his lower lip for 10 months. What is the cause? What is the treatment?
Article
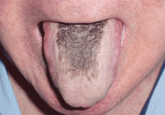
Black hairy tongue
A 71-year-old man presents for evaluation of an asymptomatic black discoloration of the tongue. What is the most likely diagnosis?
Article
Sudden hair loss associated with trachyonychia
A 30-year old woman has been having episodes of sudden hair loss in well-demarcated areas of her scalp for the past 6 months. What is the most...
Article
Generalized pruritus after a beach vacation
A 25-year-old man presents with a 2-month history of generalized itching that began 5 weeks after a trip to Brazil, during which he had sexual...
Article
Acute facial purpura in an 82-year-old woman with a respiratory tract infection
The lesions appeared suddenly and spontaneously and were not associated with trauma. What is the most likely diagnosis?