User login
Waxy Indurated Plaques on the Eyelids
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
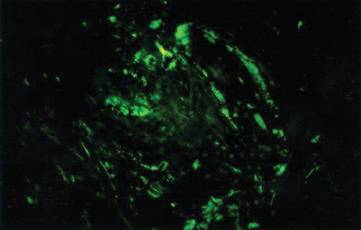
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
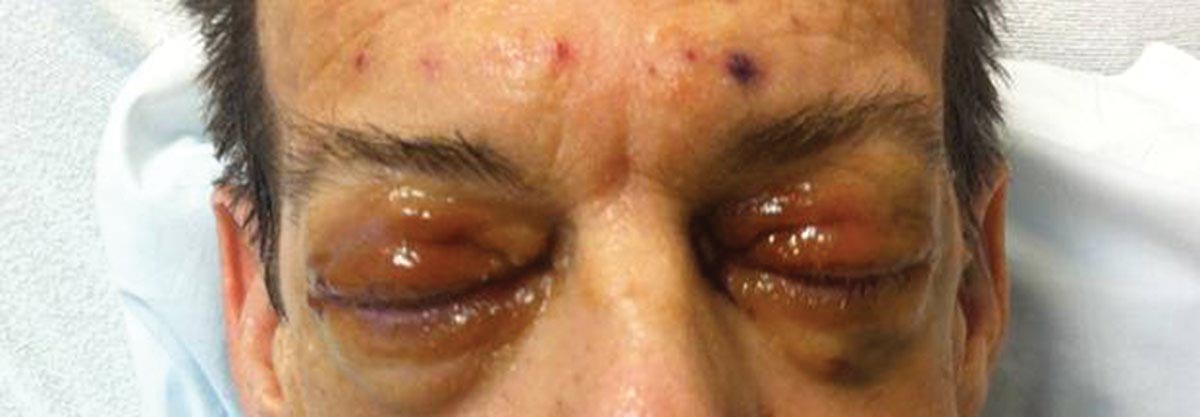
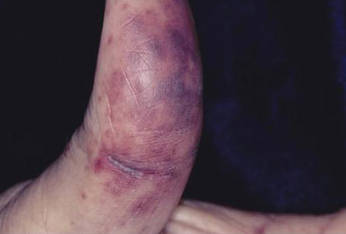
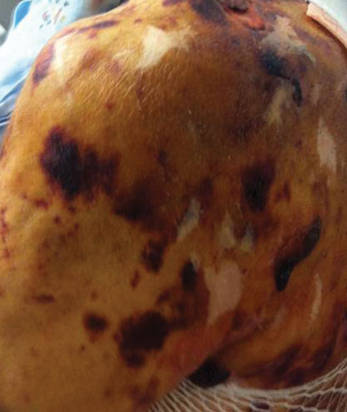
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
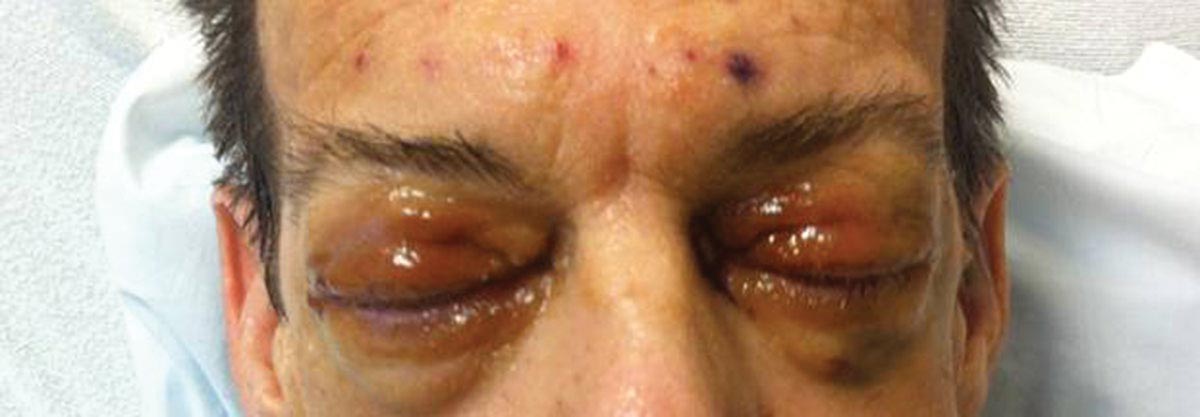
A 57-year-old man who underwent an orthotopic liver transplant 10 months prior for autoimmune hepatitis developed a rash on the trunk, arms, and legs. A dermatology consultation was requested. On evaluation, the patient was noted to have purpuric patches on the trunk and extremities, as well as prominent, bilateral, waxy and indurated periorbital plaques. The patient’s medical history revealed anemia, osteopenia, multiple bone fractures, and renal impairment.