User login
Elements of Confusion
A 23‐year‐old man presented to his family physician's office with a 2‐week history of fever, chills, night sweats, anorexia, and fatigue. This was associated with a 4‐month history of a nonproductive cough and a 20‐pound involuntary weight loss. He denied shortness of breath, chest pain, headaches, abdominal pain, vomiting, diarrhea, dysuria, and rash. There was no recent travel, sick contacts, or animal exposures.
This patient's symptoms could represent an underlying infectious, neoplastic, or inflammatory process. I would ascertain any relevant personal or family history and explore whether the patient has risk factors for human immunodeficiency virus (HIV) infection or tuberculosis (TB). On physical examination, I would listen for a heart murmur and look for lymphadenopathy, hepatosplenomegaly, and arthritis. Investigations including cultures, urinalysis, and a chest radiograph would be indicated at this time.
During the 2 weeks after his initial presentation, he experienced persistent fever, and further weight loss. He was admitted to the hospital to determine the etiology of his symptoms. The patient had no previous medical problems. On initial examination, his temperature was 102 degrees Fahrenheit, blood pressure was 100/65 mmHg, heart rate was 105 per minute, respiratory rate was 22 breaths per minute and oxygen saturation was normal on ambient air. He appeared cachectic. He was oriented to person, place, and time. Head and neck examination revealed no intraoral pathology, lymphadenopathy or scleral icterus, but did reveal conjunctival pallor. The chest was clear to auscultation, and the cardiovascular examination revealed a normal apical impulse and heart sounds with no murmurs. There was peripheral edema to the level of the mid‐shins bilaterally. The abdomen was soft and non‐tender with no appreciable hepatosplenomegaly. There were no stigmata of chronic liver disease. There was no axillary or inguinal lymphadenopathy. The remainder of the examination was normal. A complete blood count showed a hemoglobin concentration of 5.2 g/dL with a mean corpuscular volume (MCV) of 89fL, white blood cells were 1,400 cells/mm3 with an absolute neutrophil count (ANC) of 800 cells/mm3 and a platelet count of 90,000 cells/mm3 The serum sodium was 124 mmol/L, potassium 3.0 mmol/L, chloride 91 mmol/L, bicarbonate 26 mmol/L, and the creatinine 1.36 mg/dL His liver enzyme profile showed aspartate aminotransferase (AST) 68 U/L (normal <35), alanine aminotransferase (ALT) 25 U/L (normal <40), alkaline phosphatase (ALP) 210 U/L (normal <110) and a total bilirubin of 1.64 mg/dL.
The patient is clearly very unwell and requires admission to the hospital for treatment and further investigation. Emergent management includes administration of intravenous fluids to correct his electrolyte abnormalities, empiric broad spectrum antibiotics (given his relative neutropenia and fever), and a transfusion for his profound anemia. I would be very concerned that he has an underlying malignancy such as lymphoma or leukemia. Pancytopenia related to decreased cell production may be secondary to infiltration (malignant or granulomatous), infection (HIV, TB, fungal, viral), or aplasia (primary or drug‐related). Less likely etiologies include B12 or folate deficiency (unlikely given the normal MCV), systemic lupus erythematosus, paroxysmal nocturnal hemoglobinuria or cell sequestration due to hypersplenism. A history of recent exposure to drugs or toxins should be elicited. The patient's pulmonary symptoms may relate to the primary disorder or may represent an infection secondary to myelosuppression. I would want an immediate review of the peripheral blood smear, a hemolysis work‐up (drawn prior to transfusion including lactate dehydrogenase [LDH], haptoglobin, fractionated bilirubin, reticulocyte count and direct antiglobulin testing), antinuclear antibody (ANA), B12 and folate levels, imaging of the chest and blood cultures.
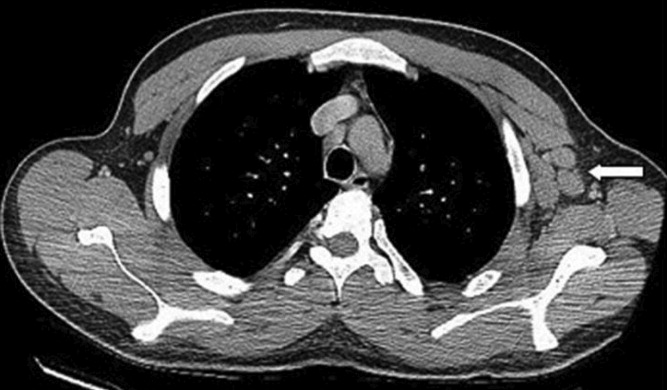
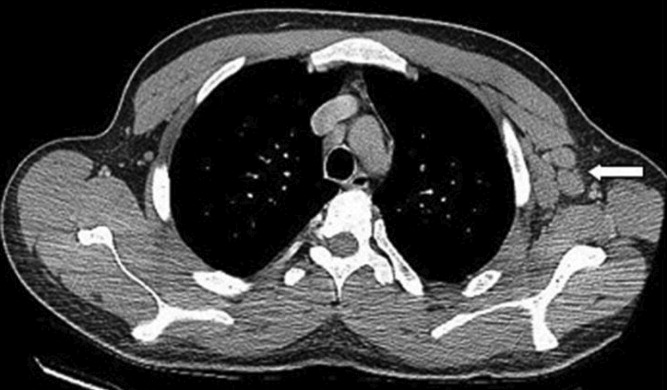
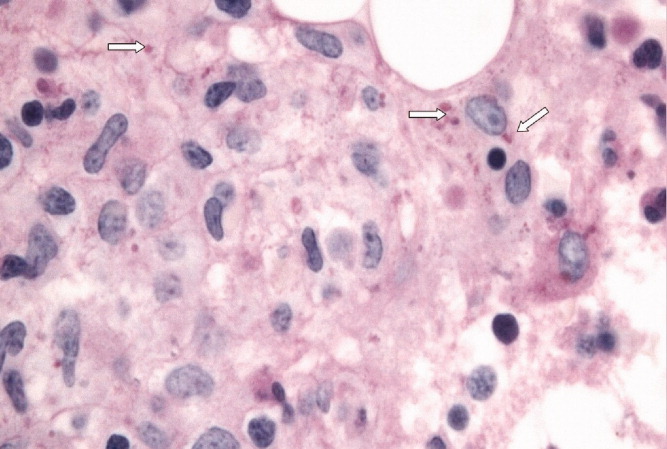
With evidence of fever and pancytopenia, acute leukemia was suspected and the patient was admitted to a hematology service. Over the next two weeks an extensive investigation including blood and urine cultures, and computed tomograms (CT) of the chest and abdomen were performed. A bone marrow aspirate and biopsy were also were done and were submitted for histopathologic examination and culture. The CT scan of the chest revealed left axillary and supraclavicular lymphadenopathy (Figure 1), and the abdominal imaging revealed splenomegaly. The blood, urine and bone marrow cultures were all negative. A peripheral blood smear showed pancytopenia with a hematologist interpretation suggesting that an intrinsic bone marrow process may be resulting in impaired cell production. The corresponding bone marrow biopsy and aspirate showed no evidence of malignancy, but there were numerous granulomata, and the periodic acid‐Schiff (PAS) and silver staining showed cells that resembled fungal elements (Figure 2).
The absence of malignant cells in the bone marrow leaves us to consider infectious and inflammatory causes of this patient's presentation. Infectious etiologies associated with bone marrow granulomata include fungal, mycobacterial, bacterial (brucellosis, typhoid and Q fever) and viral pathogens including HIV, Epstein‐Barr virus (EBV), and cytomegalovirus (CMV). Noninfectious causes include sarcoidosis, drug effects, and autoimmune conditions. The PAS and silver staining suggests this patient has a disseminated fungal infection. Disseminated Histoplasma capsulatum is the most likely organism but blastomycosis and coccidioidomycosis should be considered. HIV and occult lymphoma are considerations as is a primary immune disorder such as common variable immunodeficiency (CVID) which can present in this age group. While there is no recent travel history, it will be critical to determine where the patient currently lives and previously resided, review the medical record for prior infections and HIV risk factors, and take a thorough occupational history.
At this point, the following investigations should be undertaken: blood, sputum, and bone marrow culture; fungal and acid‐fast bacilli (AFB) stains on sputum and bone marrow; Histoplasma urine antigen; tuberculin skin test; serology for HIV and histoplasmosis; and serum protein electrophoresis with immunofixation and quantitation of immunoglobulins.
Acid fast staining of the bone marrow as well as mycobacterial and fungal cultures were negative. He lived in eastern Ontario and worked in construction. He reported helping tear down an old cabin in a wooded area, but denied any insect bites. This project coincided with the onset of his cough. He had no history of high risk sexual activity, intravenous drug use, tattoos or blood transfusions previous to his presentation. The HIV test was negative. His clinicians at this point considered a disseminated fungal infection as a cause for his symptoms and started him empirically on itraconazole He was discharged from the hospital with a plan for close outpatient followup. Within three days of discharge on the itraconazole, the patient's fever began to diminish, but did not completely resolve.
The clinical picture including cough, geography, and recent occupational exposure is entirely consistent with disseminated histoplasmosis. However, we are still lacking microbiologic confirmation of the diagnosis. Sarcoidosis and occult malignancy must still be considered. In the absence of a definitive diagnosis, I would consider bronchoscopy with bronchoalveolar lavage (BAL) and obtaining a lymph node or liver biopsy for microbiologic and pathologic examination. With the patient now receiving antifungal therapy, a diagnosis of histoplasmosis would be supported by a response to therapy, declining Histoplasma antigen levels and clinical improvement including recovery of his bone marrow.
The urine specimen was negative for Histoplasma antigen. Seven days after initiating itraconazole, he developed jaundice and confusion and was taken back to the hospital. On presentation, he was disoriented but awake. His temperature was 103.1 degrees Fahrenheit, blood pressure was 90/60 mm Hg, heart rate was 115 per minute, and oxygen saturation was normal on room air. He was obviously jaundiced, and more cachectic than previous. The neurologic examination demonstrated disorientation with no localizing findings. The chest and cardiovascular examinations were normal. His abdomen was soft and non‐tender with no evidence of hepatomegaly, but the spleen tip was palpable. There was no ascites or any other signs of portal hypertension, but his peripheral edema was worse than before and asterixis was present. The remainder of the examination was unchanged from previous. His laboratory investigations at this point showed a bilirubin of 18.5 mg/dL, AST 269 U/L, ALT 76 U/L, ALP 165 U/L, albumin 18 g/L, fibrinogen 1.53 g/L (normal 1.5‐3.5), triglycerides 2.4 mmol/L (normal <2), ferritin 59415 ug/L (normal 22‐275) an international normalized ratio (INR) of 2.65. His complete blood count still showed pancytopenia.
The patient has now developed fulminant hepatic failure. He requires volume resuscitation, drawing of repeat cultures, initiation of empiric broad spectrum antibiotics, urgent hepatology consultation and intensive care unit (ICU) support. The most common causes of acute liver failure are drug toxicity (including acetaminophen), viral hepatitis, Wilson's disease, Budd‐Chiari syndrome, cryptogenic liver disease and fatty infiltration. The critical diagnostic issue at this point is to determine if the liver failure is a secondary process (in which case drug toxicity due to itraconazole would be the most likely cause) or if this represents evolution of his primary disease with extensive hepatic involvement. Liver failure due to itraconazole has been reported and given the lack of microbiologic confirmation of a fungal infection, this agent should clearly be discontinued. Returning to our initial differential diagnosis of this man's granulomatous bone marrow infiltration and pancytopenia, etiologies which may progress to hepatic failure include viral infections (EBV or CMV) and malignancy. This patient's presentation could be an unusual manifestation of a common illness such as EBV or a rapidly progressive lymphoma. An abdominal Doppler ultrasound is required to rule out Budd‐Chiari syndrome. Given his abrupt change in clinical status, I would repeat a CT scan of his chest and abdomen to evaluate for evidence of infection, infiltration, or malignancy. Owing to the uncertainty regarding this patient's diagnosis and the rapidly progressive nature of his disease, serious consideration must be given to a transjugular liver biopsy.
Soon after admission, he developed hematemesis. He was given multiple blood transfusions, and then intravenous fluids, broad spectrum antibiotics and lactulose. Upper gastrointestinal endoscopy showed no varices, but did reveal multiple esophageal and gastric ulcerations. He was then transferred to a liver transplant center where repeat bone marrow biopsy and a liver biopsy were done. Both revealed extensive granulomatosis and the bone marrow biopsy showed evidence of hemophagocytosis (Figure 3).
The finding of hemophagocytosis in the setting of fever, hepatosplenomegaly, and pancytopenia is consistent with a diagnosis of hemophagocytic lymphohistiocytosis (HLH). The cornerstone of therapy for patients with HLH is suppression of the severe inflammatory response with corticosteroids, etoposide and cyclosporin. Patients who respond to this are candidates for allogeneic stem cell transplant with curative intent. This patient's hepatic dysfunction precludes the use of etoposide and initial therapy should therefore include dexamethasone and cyclosporin.
All bacterial, fungal, and mycobacterial cultures again demonstrated no growth. Broad spectrum antibiotics were continued, and empiric intravenous amphotericin B was added. He became hemodynamically unstable, was intubated, put on mechanical ventilation and required vasoactive medications to maintain his blood pressure. An empiric course of pulse corticosteroids was given for the possibility of sarcoidosis. His blood pressure stabilized, though he continued to require vasopressors.
While HLH has been very rarely reported in association with sarcoidosis, the underlying pathogenesis of his clinical presentation (infectious, neoplastic, or inflammatory) has not yet been confirmed. In the meantime, I would continue with supportive care and intravenous corticosteroids.
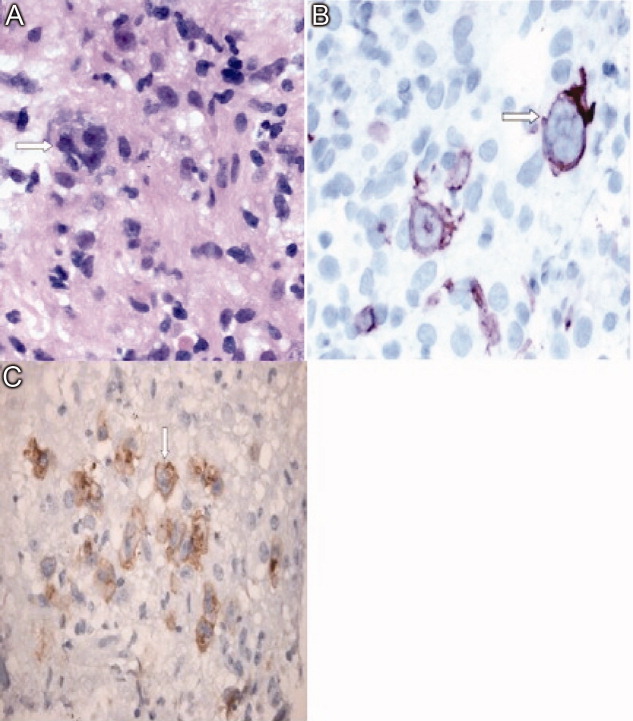
Immunohistochemical studies of the liver biopsy returned showing CD15/30+ cells with weak‐to‐negative CD45 expression cells typical of Hodgkin lymphoma (HL) (Figure 4). He was started on chemotherapy, but over 48 hours became progressively more hypotensive. The patient died of Klebsiella and Pseudomonas sepsis on the 7th hospital day. Post‐mortem immunohistochemical examination revealed evidence of Hodgkin disease in the axillary lymph nodes, bone marrow and liver. The bone marrow showed evidence of hemophagocytosis and was also positive for Epstein‐Barr encoded RNA (EBER). Serologic studies were subsequently available and revealed positive EBV IgM against the viral capsid antigen (VCA) as well as EBV IgG VCA, which in conjunction with the marrow findings, was highly suggestive of reactivation EBV disease.
Discussion
This patient's diagnostic course led both the clinical team and discussant down a winding path, which ultimately ended in the finding of Hodgkin lymphoma, a relatively common diagnosis that had been clouded by seemingly contradictory clinical and laboratory data. The provisional diagnosis of disseminated histoplasmosis was reasonable given that H. capsulatum is endemic in Ontario and that the patient's occupation placed him at risk of infection. Given the acuity of his illness, empiric antifungal therapy based on the report of fungal elements on bone marrow examination seemed reasonable. However, Histoplasma urinary antigen testing has been shown in the literature to be 98% sensitive in immunosuppressed populations, and the negative result prompted a re‐examination of the marrow specimen. The previously described fungal elements were felt to be most likely artifact, and the underlying diagnosis was reconsidered.1 This is when the repeat bone marrow examination pointed towards the diagnosis of HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a severe, systemic hyperinflammatory disorder characterized by histiocytic proliferation that may be primary or can be triggered by infection, connective tissue diseases or malignancy.25 The central pathogenesis involves dysregulated Th1 cytokine secretion. This results in an uncontrolled accumulation of activated T‐lymphocytes and histiocytes in various organs including the liver, spleen and bone marrow. The infiltration of histiocytes into major organs can lead to disruption of function and multiorgan failure.6 Viruses are the most common infectious triggers of HLH, particularly EBV, and lymphoma is the most common associated malignancy.25 It is hypothesized that EBV can interfere with normal lymphocyte signaling pathways leading to the aforementioned over‐expression of Th1 cytokines, which can then trigger HLH.7 The diagnosis of HLH is based on a combination of clinical and laboratory parameters as outlined in Table 1.8 Our patient met all five of the major criteria.
| |
| Major criteria | 1. Fever |
| 2. Splenomegaly | |
| 3. Cytopenia in two or more cell lines | |
| 4. Hypertriglyceridemia or hypofibrinogenemia | |
| 5. Hemophagocytosis on histopathologic examination | |
| Alternative criteria | A. Low or absent natural killer cell activity |
| B. Serum ferritin level >500 ug/L | |
| C. Soluble CD‐25 level >2400 U/mL | |
The recommended treatment of HLH involves the administration of the HLH‐94 protocol consisting of corticosteroids, cyclosporine and etoposide.9, 10 Those who survive this initial phase are recommended for hematopoetic stem cell transplantation producing an overall 3‐year survival rate of 64%. However, those who do not receive early etoposide therapy fare much worse, with a mortality rate of 92%.10 This patient was not able to receive etoposide because of his decompensated liver disease.
In this case, the development of Hodgkin lymphoma involving the bone marrow and liver may have resulted in a state of immune suppression. The loss of immune function likely allowed the reactivation of Epstein‐Barr virus which triggered HLH and his fulminant presentation.35,9 Indeed, both the liver and bone marrow samples showed evidence of EBV reactivation as evidenced by the presence of EBER. The diagnosis of Hodgkin lymphoma was made from a liver biopsy specimen rather than bone marrow examination. The diagnosis of Hodgkin lymphoma is based on the presence of Reed‐Sternberg cells surrounded by an inflammatory milieu of cells including variable numbers of small lymphocytes, neutrophils eosinophils and fibroblasts. The HLH‐induced pancytopenia depleted the aforementioned inflammatory milieu in the bone marrow, which obscured the diagnosis of Hodgkin lymphoma. Unfortunately, lymphoma‐associated HLH has a very poor prognosis with a mortality rate of up to 60%.4 At the outset of this case, the reported fungal elements proved to be a source of confusion which delayed the diagnosis of Hodgkin lymphoma. Given the poor prognosis of lymphoma‐associated HLH, it is unlikely this would have had any effect on the ultimate outcome.
Teaching Points
-
HLH is a rare and complex hyperinflammatory disorder which may present as pancytopenia.
-
Triggers of HLH can include infections (particularly EBV), malignancy (particularly lymphoma) and connective tissue diseases.
-
The diagnosis of HLH is based on clinical and laboratory criteria, including cytopenias that may make the evaluation for triggering conditions such as HL more difficult.
-
Lymphoma should be included in the differential diagnosis of granulomatous inflammation.
Acknowledgements
The authors acknowledge Ralph Meyer, MD (Queen's University, Kingston, Ontario) for his comments on a draft of this paper and Drs. David Barth and Maha Guindi (Department of Laboratory Medicine, University Health Network) for their reviews of the pathology specimens.
- ,Antigen detection for diagnosis of the endemic mycoses.Curr Fung Infect Rep.2008;4:189–193.
- ,,, et al.Lymphoma‐associated hemophagocytic syndrome: Clinical features and treatment outcome.Ann Hematol.2007;86:493–498.
- ,,, et al.Hodgkin lymphoma‐associated hemophagocytic syndrome: A disorder strongly correlated with Epstein‐Barr virus.Clin Inf Dis.2008;47:531–534.
- ,.Hemophagocytic syndrome associated with Hodgkin lymphoma and pneumocystis jiroveci pneumonitis.Br J Hematol.2007;138:672.
- ,,, et al.Infections associated with haemophagocytic syndrome.Lancet Infect Dis.2007;12:814–822.
- ,,,.Hemophagocytic lymphohistiocytic syndrome: Unrecognized cause of multiple organ failure.Pediatr Crit Care Med.2000;1:51–54.
- ,,, et al.Epstein‐Barr virus LMP1 Inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome.Blood.2005;106:3090–3096.
- ,,.Diagnostic guidelines for hemophagocytic lymphohistiocytosis.Semin Oncol.1991;18:29–33
- ,,, et al.Treatment of Epstein‐Barr virus‐associated hemophagocytic lymphohistiocytosis (EBV‐HLH) in young adults: A report from the HLH Study Center.Med Pediatr Oncol.2003;41:103–109.
- ,,, et al.Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis.Br J Haematol.2005;129:622–630.
A 23‐year‐old man presented to his family physician's office with a 2‐week history of fever, chills, night sweats, anorexia, and fatigue. This was associated with a 4‐month history of a nonproductive cough and a 20‐pound involuntary weight loss. He denied shortness of breath, chest pain, headaches, abdominal pain, vomiting, diarrhea, dysuria, and rash. There was no recent travel, sick contacts, or animal exposures.
This patient's symptoms could represent an underlying infectious, neoplastic, or inflammatory process. I would ascertain any relevant personal or family history and explore whether the patient has risk factors for human immunodeficiency virus (HIV) infection or tuberculosis (TB). On physical examination, I would listen for a heart murmur and look for lymphadenopathy, hepatosplenomegaly, and arthritis. Investigations including cultures, urinalysis, and a chest radiograph would be indicated at this time.
During the 2 weeks after his initial presentation, he experienced persistent fever, and further weight loss. He was admitted to the hospital to determine the etiology of his symptoms. The patient had no previous medical problems. On initial examination, his temperature was 102 degrees Fahrenheit, blood pressure was 100/65 mmHg, heart rate was 105 per minute, respiratory rate was 22 breaths per minute and oxygen saturation was normal on ambient air. He appeared cachectic. He was oriented to person, place, and time. Head and neck examination revealed no intraoral pathology, lymphadenopathy or scleral icterus, but did reveal conjunctival pallor. The chest was clear to auscultation, and the cardiovascular examination revealed a normal apical impulse and heart sounds with no murmurs. There was peripheral edema to the level of the mid‐shins bilaterally. The abdomen was soft and non‐tender with no appreciable hepatosplenomegaly. There were no stigmata of chronic liver disease. There was no axillary or inguinal lymphadenopathy. The remainder of the examination was normal. A complete blood count showed a hemoglobin concentration of 5.2 g/dL with a mean corpuscular volume (MCV) of 89fL, white blood cells were 1,400 cells/mm3 with an absolute neutrophil count (ANC) of 800 cells/mm3 and a platelet count of 90,000 cells/mm3 The serum sodium was 124 mmol/L, potassium 3.0 mmol/L, chloride 91 mmol/L, bicarbonate 26 mmol/L, and the creatinine 1.36 mg/dL His liver enzyme profile showed aspartate aminotransferase (AST) 68 U/L (normal <35), alanine aminotransferase (ALT) 25 U/L (normal <40), alkaline phosphatase (ALP) 210 U/L (normal <110) and a total bilirubin of 1.64 mg/dL.
The patient is clearly very unwell and requires admission to the hospital for treatment and further investigation. Emergent management includes administration of intravenous fluids to correct his electrolyte abnormalities, empiric broad spectrum antibiotics (given his relative neutropenia and fever), and a transfusion for his profound anemia. I would be very concerned that he has an underlying malignancy such as lymphoma or leukemia. Pancytopenia related to decreased cell production may be secondary to infiltration (malignant or granulomatous), infection (HIV, TB, fungal, viral), or aplasia (primary or drug‐related). Less likely etiologies include B12 or folate deficiency (unlikely given the normal MCV), systemic lupus erythematosus, paroxysmal nocturnal hemoglobinuria or cell sequestration due to hypersplenism. A history of recent exposure to drugs or toxins should be elicited. The patient's pulmonary symptoms may relate to the primary disorder or may represent an infection secondary to myelosuppression. I would want an immediate review of the peripheral blood smear, a hemolysis work‐up (drawn prior to transfusion including lactate dehydrogenase [LDH], haptoglobin, fractionated bilirubin, reticulocyte count and direct antiglobulin testing), antinuclear antibody (ANA), B12 and folate levels, imaging of the chest and blood cultures.
With evidence of fever and pancytopenia, acute leukemia was suspected and the patient was admitted to a hematology service. Over the next two weeks an extensive investigation including blood and urine cultures, and computed tomograms (CT) of the chest and abdomen were performed. A bone marrow aspirate and biopsy were also were done and were submitted for histopathologic examination and culture. The CT scan of the chest revealed left axillary and supraclavicular lymphadenopathy (Figure 1), and the abdominal imaging revealed splenomegaly. The blood, urine and bone marrow cultures were all negative. A peripheral blood smear showed pancytopenia with a hematologist interpretation suggesting that an intrinsic bone marrow process may be resulting in impaired cell production. The corresponding bone marrow biopsy and aspirate showed no evidence of malignancy, but there were numerous granulomata, and the periodic acid‐Schiff (PAS) and silver staining showed cells that resembled fungal elements (Figure 2).
The absence of malignant cells in the bone marrow leaves us to consider infectious and inflammatory causes of this patient's presentation. Infectious etiologies associated with bone marrow granulomata include fungal, mycobacterial, bacterial (brucellosis, typhoid and Q fever) and viral pathogens including HIV, Epstein‐Barr virus (EBV), and cytomegalovirus (CMV). Noninfectious causes include sarcoidosis, drug effects, and autoimmune conditions. The PAS and silver staining suggests this patient has a disseminated fungal infection. Disseminated Histoplasma capsulatum is the most likely organism but blastomycosis and coccidioidomycosis should be considered. HIV and occult lymphoma are considerations as is a primary immune disorder such as common variable immunodeficiency (CVID) which can present in this age group. While there is no recent travel history, it will be critical to determine where the patient currently lives and previously resided, review the medical record for prior infections and HIV risk factors, and take a thorough occupational history.
At this point, the following investigations should be undertaken: blood, sputum, and bone marrow culture; fungal and acid‐fast bacilli (AFB) stains on sputum and bone marrow; Histoplasma urine antigen; tuberculin skin test; serology for HIV and histoplasmosis; and serum protein electrophoresis with immunofixation and quantitation of immunoglobulins.
Acid fast staining of the bone marrow as well as mycobacterial and fungal cultures were negative. He lived in eastern Ontario and worked in construction. He reported helping tear down an old cabin in a wooded area, but denied any insect bites. This project coincided with the onset of his cough. He had no history of high risk sexual activity, intravenous drug use, tattoos or blood transfusions previous to his presentation. The HIV test was negative. His clinicians at this point considered a disseminated fungal infection as a cause for his symptoms and started him empirically on itraconazole He was discharged from the hospital with a plan for close outpatient followup. Within three days of discharge on the itraconazole, the patient's fever began to diminish, but did not completely resolve.
The clinical picture including cough, geography, and recent occupational exposure is entirely consistent with disseminated histoplasmosis. However, we are still lacking microbiologic confirmation of the diagnosis. Sarcoidosis and occult malignancy must still be considered. In the absence of a definitive diagnosis, I would consider bronchoscopy with bronchoalveolar lavage (BAL) and obtaining a lymph node or liver biopsy for microbiologic and pathologic examination. With the patient now receiving antifungal therapy, a diagnosis of histoplasmosis would be supported by a response to therapy, declining Histoplasma antigen levels and clinical improvement including recovery of his bone marrow.
The urine specimen was negative for Histoplasma antigen. Seven days after initiating itraconazole, he developed jaundice and confusion and was taken back to the hospital. On presentation, he was disoriented but awake. His temperature was 103.1 degrees Fahrenheit, blood pressure was 90/60 mm Hg, heart rate was 115 per minute, and oxygen saturation was normal on room air. He was obviously jaundiced, and more cachectic than previous. The neurologic examination demonstrated disorientation with no localizing findings. The chest and cardiovascular examinations were normal. His abdomen was soft and non‐tender with no evidence of hepatomegaly, but the spleen tip was palpable. There was no ascites or any other signs of portal hypertension, but his peripheral edema was worse than before and asterixis was present. The remainder of the examination was unchanged from previous. His laboratory investigations at this point showed a bilirubin of 18.5 mg/dL, AST 269 U/L, ALT 76 U/L, ALP 165 U/L, albumin 18 g/L, fibrinogen 1.53 g/L (normal 1.5‐3.5), triglycerides 2.4 mmol/L (normal <2), ferritin 59415 ug/L (normal 22‐275) an international normalized ratio (INR) of 2.65. His complete blood count still showed pancytopenia.
The patient has now developed fulminant hepatic failure. He requires volume resuscitation, drawing of repeat cultures, initiation of empiric broad spectrum antibiotics, urgent hepatology consultation and intensive care unit (ICU) support. The most common causes of acute liver failure are drug toxicity (including acetaminophen), viral hepatitis, Wilson's disease, Budd‐Chiari syndrome, cryptogenic liver disease and fatty infiltration. The critical diagnostic issue at this point is to determine if the liver failure is a secondary process (in which case drug toxicity due to itraconazole would be the most likely cause) or if this represents evolution of his primary disease with extensive hepatic involvement. Liver failure due to itraconazole has been reported and given the lack of microbiologic confirmation of a fungal infection, this agent should clearly be discontinued. Returning to our initial differential diagnosis of this man's granulomatous bone marrow infiltration and pancytopenia, etiologies which may progress to hepatic failure include viral infections (EBV or CMV) and malignancy. This patient's presentation could be an unusual manifestation of a common illness such as EBV or a rapidly progressive lymphoma. An abdominal Doppler ultrasound is required to rule out Budd‐Chiari syndrome. Given his abrupt change in clinical status, I would repeat a CT scan of his chest and abdomen to evaluate for evidence of infection, infiltration, or malignancy. Owing to the uncertainty regarding this patient's diagnosis and the rapidly progressive nature of his disease, serious consideration must be given to a transjugular liver biopsy.
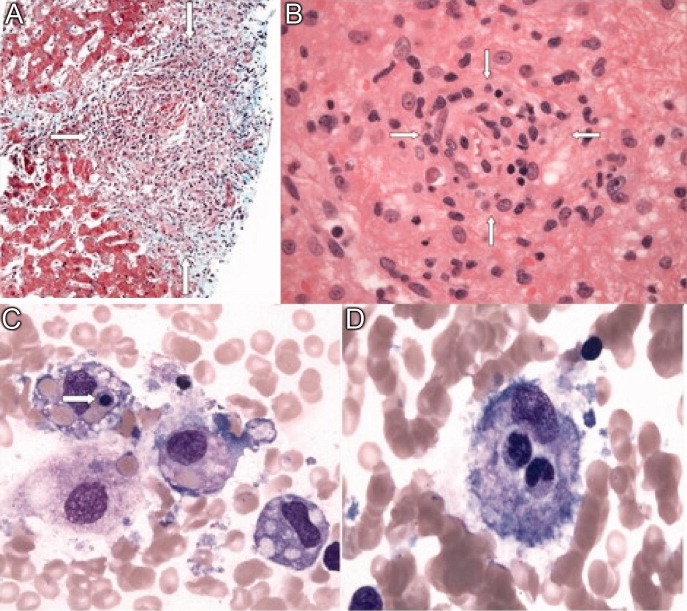
Soon after admission, he developed hematemesis. He was given multiple blood transfusions, and then intravenous fluids, broad spectrum antibiotics and lactulose. Upper gastrointestinal endoscopy showed no varices, but did reveal multiple esophageal and gastric ulcerations. He was then transferred to a liver transplant center where repeat bone marrow biopsy and a liver biopsy were done. Both revealed extensive granulomatosis and the bone marrow biopsy showed evidence of hemophagocytosis (Figure 3).
The finding of hemophagocytosis in the setting of fever, hepatosplenomegaly, and pancytopenia is consistent with a diagnosis of hemophagocytic lymphohistiocytosis (HLH). The cornerstone of therapy for patients with HLH is suppression of the severe inflammatory response with corticosteroids, etoposide and cyclosporin. Patients who respond to this are candidates for allogeneic stem cell transplant with curative intent. This patient's hepatic dysfunction precludes the use of etoposide and initial therapy should therefore include dexamethasone and cyclosporin.
All bacterial, fungal, and mycobacterial cultures again demonstrated no growth. Broad spectrum antibiotics were continued, and empiric intravenous amphotericin B was added. He became hemodynamically unstable, was intubated, put on mechanical ventilation and required vasoactive medications to maintain his blood pressure. An empiric course of pulse corticosteroids was given for the possibility of sarcoidosis. His blood pressure stabilized, though he continued to require vasopressors.
While HLH has been very rarely reported in association with sarcoidosis, the underlying pathogenesis of his clinical presentation (infectious, neoplastic, or inflammatory) has not yet been confirmed. In the meantime, I would continue with supportive care and intravenous corticosteroids.
Immunohistochemical studies of the liver biopsy returned showing CD15/30+ cells with weak‐to‐negative CD45 expression cells typical of Hodgkin lymphoma (HL) (Figure 4). He was started on chemotherapy, but over 48 hours became progressively more hypotensive. The patient died of Klebsiella and Pseudomonas sepsis on the 7th hospital day. Post‐mortem immunohistochemical examination revealed evidence of Hodgkin disease in the axillary lymph nodes, bone marrow and liver. The bone marrow showed evidence of hemophagocytosis and was also positive for Epstein‐Barr encoded RNA (EBER). Serologic studies were subsequently available and revealed positive EBV IgM against the viral capsid antigen (VCA) as well as EBV IgG VCA, which in conjunction with the marrow findings, was highly suggestive of reactivation EBV disease.
Discussion
This patient's diagnostic course led both the clinical team and discussant down a winding path, which ultimately ended in the finding of Hodgkin lymphoma, a relatively common diagnosis that had been clouded by seemingly contradictory clinical and laboratory data. The provisional diagnosis of disseminated histoplasmosis was reasonable given that H. capsulatum is endemic in Ontario and that the patient's occupation placed him at risk of infection. Given the acuity of his illness, empiric antifungal therapy based on the report of fungal elements on bone marrow examination seemed reasonable. However, Histoplasma urinary antigen testing has been shown in the literature to be 98% sensitive in immunosuppressed populations, and the negative result prompted a re‐examination of the marrow specimen. The previously described fungal elements were felt to be most likely artifact, and the underlying diagnosis was reconsidered.1 This is when the repeat bone marrow examination pointed towards the diagnosis of HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a severe, systemic hyperinflammatory disorder characterized by histiocytic proliferation that may be primary or can be triggered by infection, connective tissue diseases or malignancy.25 The central pathogenesis involves dysregulated Th1 cytokine secretion. This results in an uncontrolled accumulation of activated T‐lymphocytes and histiocytes in various organs including the liver, spleen and bone marrow. The infiltration of histiocytes into major organs can lead to disruption of function and multiorgan failure.6 Viruses are the most common infectious triggers of HLH, particularly EBV, and lymphoma is the most common associated malignancy.25 It is hypothesized that EBV can interfere with normal lymphocyte signaling pathways leading to the aforementioned over‐expression of Th1 cytokines, which can then trigger HLH.7 The diagnosis of HLH is based on a combination of clinical and laboratory parameters as outlined in Table 1.8 Our patient met all five of the major criteria.
| |
| Major criteria | 1. Fever |
| 2. Splenomegaly | |
| 3. Cytopenia in two or more cell lines | |
| 4. Hypertriglyceridemia or hypofibrinogenemia | |
| 5. Hemophagocytosis on histopathologic examination | |
| Alternative criteria | A. Low or absent natural killer cell activity |
| B. Serum ferritin level >500 ug/L | |
| C. Soluble CD‐25 level >2400 U/mL | |
The recommended treatment of HLH involves the administration of the HLH‐94 protocol consisting of corticosteroids, cyclosporine and etoposide.9, 10 Those who survive this initial phase are recommended for hematopoetic stem cell transplantation producing an overall 3‐year survival rate of 64%. However, those who do not receive early etoposide therapy fare much worse, with a mortality rate of 92%.10 This patient was not able to receive etoposide because of his decompensated liver disease.
In this case, the development of Hodgkin lymphoma involving the bone marrow and liver may have resulted in a state of immune suppression. The loss of immune function likely allowed the reactivation of Epstein‐Barr virus which triggered HLH and his fulminant presentation.35,9 Indeed, both the liver and bone marrow samples showed evidence of EBV reactivation as evidenced by the presence of EBER. The diagnosis of Hodgkin lymphoma was made from a liver biopsy specimen rather than bone marrow examination. The diagnosis of Hodgkin lymphoma is based on the presence of Reed‐Sternberg cells surrounded by an inflammatory milieu of cells including variable numbers of small lymphocytes, neutrophils eosinophils and fibroblasts. The HLH‐induced pancytopenia depleted the aforementioned inflammatory milieu in the bone marrow, which obscured the diagnosis of Hodgkin lymphoma. Unfortunately, lymphoma‐associated HLH has a very poor prognosis with a mortality rate of up to 60%.4 At the outset of this case, the reported fungal elements proved to be a source of confusion which delayed the diagnosis of Hodgkin lymphoma. Given the poor prognosis of lymphoma‐associated HLH, it is unlikely this would have had any effect on the ultimate outcome.
Teaching Points
-
HLH is a rare and complex hyperinflammatory disorder which may present as pancytopenia.
-
Triggers of HLH can include infections (particularly EBV), malignancy (particularly lymphoma) and connective tissue diseases.
-
The diagnosis of HLH is based on clinical and laboratory criteria, including cytopenias that may make the evaluation for triggering conditions such as HL more difficult.
-
Lymphoma should be included in the differential diagnosis of granulomatous inflammation.
Acknowledgements
The authors acknowledge Ralph Meyer, MD (Queen's University, Kingston, Ontario) for his comments on a draft of this paper and Drs. David Barth and Maha Guindi (Department of Laboratory Medicine, University Health Network) for their reviews of the pathology specimens.
A 23‐year‐old man presented to his family physician's office with a 2‐week history of fever, chills, night sweats, anorexia, and fatigue. This was associated with a 4‐month history of a nonproductive cough and a 20‐pound involuntary weight loss. He denied shortness of breath, chest pain, headaches, abdominal pain, vomiting, diarrhea, dysuria, and rash. There was no recent travel, sick contacts, or animal exposures.
This patient's symptoms could represent an underlying infectious, neoplastic, or inflammatory process. I would ascertain any relevant personal or family history and explore whether the patient has risk factors for human immunodeficiency virus (HIV) infection or tuberculosis (TB). On physical examination, I would listen for a heart murmur and look for lymphadenopathy, hepatosplenomegaly, and arthritis. Investigations including cultures, urinalysis, and a chest radiograph would be indicated at this time.
During the 2 weeks after his initial presentation, he experienced persistent fever, and further weight loss. He was admitted to the hospital to determine the etiology of his symptoms. The patient had no previous medical problems. On initial examination, his temperature was 102 degrees Fahrenheit, blood pressure was 100/65 mmHg, heart rate was 105 per minute, respiratory rate was 22 breaths per minute and oxygen saturation was normal on ambient air. He appeared cachectic. He was oriented to person, place, and time. Head and neck examination revealed no intraoral pathology, lymphadenopathy or scleral icterus, but did reveal conjunctival pallor. The chest was clear to auscultation, and the cardiovascular examination revealed a normal apical impulse and heart sounds with no murmurs. There was peripheral edema to the level of the mid‐shins bilaterally. The abdomen was soft and non‐tender with no appreciable hepatosplenomegaly. There were no stigmata of chronic liver disease. There was no axillary or inguinal lymphadenopathy. The remainder of the examination was normal. A complete blood count showed a hemoglobin concentration of 5.2 g/dL with a mean corpuscular volume (MCV) of 89fL, white blood cells were 1,400 cells/mm3 with an absolute neutrophil count (ANC) of 800 cells/mm3 and a platelet count of 90,000 cells/mm3 The serum sodium was 124 mmol/L, potassium 3.0 mmol/L, chloride 91 mmol/L, bicarbonate 26 mmol/L, and the creatinine 1.36 mg/dL His liver enzyme profile showed aspartate aminotransferase (AST) 68 U/L (normal <35), alanine aminotransferase (ALT) 25 U/L (normal <40), alkaline phosphatase (ALP) 210 U/L (normal <110) and a total bilirubin of 1.64 mg/dL.
The patient is clearly very unwell and requires admission to the hospital for treatment and further investigation. Emergent management includes administration of intravenous fluids to correct his electrolyte abnormalities, empiric broad spectrum antibiotics (given his relative neutropenia and fever), and a transfusion for his profound anemia. I would be very concerned that he has an underlying malignancy such as lymphoma or leukemia. Pancytopenia related to decreased cell production may be secondary to infiltration (malignant or granulomatous), infection (HIV, TB, fungal, viral), or aplasia (primary or drug‐related). Less likely etiologies include B12 or folate deficiency (unlikely given the normal MCV), systemic lupus erythematosus, paroxysmal nocturnal hemoglobinuria or cell sequestration due to hypersplenism. A history of recent exposure to drugs or toxins should be elicited. The patient's pulmonary symptoms may relate to the primary disorder or may represent an infection secondary to myelosuppression. I would want an immediate review of the peripheral blood smear, a hemolysis work‐up (drawn prior to transfusion including lactate dehydrogenase [LDH], haptoglobin, fractionated bilirubin, reticulocyte count and direct antiglobulin testing), antinuclear antibody (ANA), B12 and folate levels, imaging of the chest and blood cultures.
With evidence of fever and pancytopenia, acute leukemia was suspected and the patient was admitted to a hematology service. Over the next two weeks an extensive investigation including blood and urine cultures, and computed tomograms (CT) of the chest and abdomen were performed. A bone marrow aspirate and biopsy were also were done and were submitted for histopathologic examination and culture. The CT scan of the chest revealed left axillary and supraclavicular lymphadenopathy (Figure 1), and the abdominal imaging revealed splenomegaly. The blood, urine and bone marrow cultures were all negative. A peripheral blood smear showed pancytopenia with a hematologist interpretation suggesting that an intrinsic bone marrow process may be resulting in impaired cell production. The corresponding bone marrow biopsy and aspirate showed no evidence of malignancy, but there were numerous granulomata, and the periodic acid‐Schiff (PAS) and silver staining showed cells that resembled fungal elements (Figure 2).
The absence of malignant cells in the bone marrow leaves us to consider infectious and inflammatory causes of this patient's presentation. Infectious etiologies associated with bone marrow granulomata include fungal, mycobacterial, bacterial (brucellosis, typhoid and Q fever) and viral pathogens including HIV, Epstein‐Barr virus (EBV), and cytomegalovirus (CMV). Noninfectious causes include sarcoidosis, drug effects, and autoimmune conditions. The PAS and silver staining suggests this patient has a disseminated fungal infection. Disseminated Histoplasma capsulatum is the most likely organism but blastomycosis and coccidioidomycosis should be considered. HIV and occult lymphoma are considerations as is a primary immune disorder such as common variable immunodeficiency (CVID) which can present in this age group. While there is no recent travel history, it will be critical to determine where the patient currently lives and previously resided, review the medical record for prior infections and HIV risk factors, and take a thorough occupational history.
At this point, the following investigations should be undertaken: blood, sputum, and bone marrow culture; fungal and acid‐fast bacilli (AFB) stains on sputum and bone marrow; Histoplasma urine antigen; tuberculin skin test; serology for HIV and histoplasmosis; and serum protein electrophoresis with immunofixation and quantitation of immunoglobulins.
Acid fast staining of the bone marrow as well as mycobacterial and fungal cultures were negative. He lived in eastern Ontario and worked in construction. He reported helping tear down an old cabin in a wooded area, but denied any insect bites. This project coincided with the onset of his cough. He had no history of high risk sexual activity, intravenous drug use, tattoos or blood transfusions previous to his presentation. The HIV test was negative. His clinicians at this point considered a disseminated fungal infection as a cause for his symptoms and started him empirically on itraconazole He was discharged from the hospital with a plan for close outpatient followup. Within three days of discharge on the itraconazole, the patient's fever began to diminish, but did not completely resolve.
The clinical picture including cough, geography, and recent occupational exposure is entirely consistent with disseminated histoplasmosis. However, we are still lacking microbiologic confirmation of the diagnosis. Sarcoidosis and occult malignancy must still be considered. In the absence of a definitive diagnosis, I would consider bronchoscopy with bronchoalveolar lavage (BAL) and obtaining a lymph node or liver biopsy for microbiologic and pathologic examination. With the patient now receiving antifungal therapy, a diagnosis of histoplasmosis would be supported by a response to therapy, declining Histoplasma antigen levels and clinical improvement including recovery of his bone marrow.
The urine specimen was negative for Histoplasma antigen. Seven days after initiating itraconazole, he developed jaundice and confusion and was taken back to the hospital. On presentation, he was disoriented but awake. His temperature was 103.1 degrees Fahrenheit, blood pressure was 90/60 mm Hg, heart rate was 115 per minute, and oxygen saturation was normal on room air. He was obviously jaundiced, and more cachectic than previous. The neurologic examination demonstrated disorientation with no localizing findings. The chest and cardiovascular examinations were normal. His abdomen was soft and non‐tender with no evidence of hepatomegaly, but the spleen tip was palpable. There was no ascites or any other signs of portal hypertension, but his peripheral edema was worse than before and asterixis was present. The remainder of the examination was unchanged from previous. His laboratory investigations at this point showed a bilirubin of 18.5 mg/dL, AST 269 U/L, ALT 76 U/L, ALP 165 U/L, albumin 18 g/L, fibrinogen 1.53 g/L (normal 1.5‐3.5), triglycerides 2.4 mmol/L (normal <2), ferritin 59415 ug/L (normal 22‐275) an international normalized ratio (INR) of 2.65. His complete blood count still showed pancytopenia.
The patient has now developed fulminant hepatic failure. He requires volume resuscitation, drawing of repeat cultures, initiation of empiric broad spectrum antibiotics, urgent hepatology consultation and intensive care unit (ICU) support. The most common causes of acute liver failure are drug toxicity (including acetaminophen), viral hepatitis, Wilson's disease, Budd‐Chiari syndrome, cryptogenic liver disease and fatty infiltration. The critical diagnostic issue at this point is to determine if the liver failure is a secondary process (in which case drug toxicity due to itraconazole would be the most likely cause) or if this represents evolution of his primary disease with extensive hepatic involvement. Liver failure due to itraconazole has been reported and given the lack of microbiologic confirmation of a fungal infection, this agent should clearly be discontinued. Returning to our initial differential diagnosis of this man's granulomatous bone marrow infiltration and pancytopenia, etiologies which may progress to hepatic failure include viral infections (EBV or CMV) and malignancy. This patient's presentation could be an unusual manifestation of a common illness such as EBV or a rapidly progressive lymphoma. An abdominal Doppler ultrasound is required to rule out Budd‐Chiari syndrome. Given his abrupt change in clinical status, I would repeat a CT scan of his chest and abdomen to evaluate for evidence of infection, infiltration, or malignancy. Owing to the uncertainty regarding this patient's diagnosis and the rapidly progressive nature of his disease, serious consideration must be given to a transjugular liver biopsy.
Soon after admission, he developed hematemesis. He was given multiple blood transfusions, and then intravenous fluids, broad spectrum antibiotics and lactulose. Upper gastrointestinal endoscopy showed no varices, but did reveal multiple esophageal and gastric ulcerations. He was then transferred to a liver transplant center where repeat bone marrow biopsy and a liver biopsy were done. Both revealed extensive granulomatosis and the bone marrow biopsy showed evidence of hemophagocytosis (Figure 3).
The finding of hemophagocytosis in the setting of fever, hepatosplenomegaly, and pancytopenia is consistent with a diagnosis of hemophagocytic lymphohistiocytosis (HLH). The cornerstone of therapy for patients with HLH is suppression of the severe inflammatory response with corticosteroids, etoposide and cyclosporin. Patients who respond to this are candidates for allogeneic stem cell transplant with curative intent. This patient's hepatic dysfunction precludes the use of etoposide and initial therapy should therefore include dexamethasone and cyclosporin.
All bacterial, fungal, and mycobacterial cultures again demonstrated no growth. Broad spectrum antibiotics were continued, and empiric intravenous amphotericin B was added. He became hemodynamically unstable, was intubated, put on mechanical ventilation and required vasoactive medications to maintain his blood pressure. An empiric course of pulse corticosteroids was given for the possibility of sarcoidosis. His blood pressure stabilized, though he continued to require vasopressors.
While HLH has been very rarely reported in association with sarcoidosis, the underlying pathogenesis of his clinical presentation (infectious, neoplastic, or inflammatory) has not yet been confirmed. In the meantime, I would continue with supportive care and intravenous corticosteroids.
Immunohistochemical studies of the liver biopsy returned showing CD15/30+ cells with weak‐to‐negative CD45 expression cells typical of Hodgkin lymphoma (HL) (Figure 4). He was started on chemotherapy, but over 48 hours became progressively more hypotensive. The patient died of Klebsiella and Pseudomonas sepsis on the 7th hospital day. Post‐mortem immunohistochemical examination revealed evidence of Hodgkin disease in the axillary lymph nodes, bone marrow and liver. The bone marrow showed evidence of hemophagocytosis and was also positive for Epstein‐Barr encoded RNA (EBER). Serologic studies were subsequently available and revealed positive EBV IgM against the viral capsid antigen (VCA) as well as EBV IgG VCA, which in conjunction with the marrow findings, was highly suggestive of reactivation EBV disease.
Discussion
This patient's diagnostic course led both the clinical team and discussant down a winding path, which ultimately ended in the finding of Hodgkin lymphoma, a relatively common diagnosis that had been clouded by seemingly contradictory clinical and laboratory data. The provisional diagnosis of disseminated histoplasmosis was reasonable given that H. capsulatum is endemic in Ontario and that the patient's occupation placed him at risk of infection. Given the acuity of his illness, empiric antifungal therapy based on the report of fungal elements on bone marrow examination seemed reasonable. However, Histoplasma urinary antigen testing has been shown in the literature to be 98% sensitive in immunosuppressed populations, and the negative result prompted a re‐examination of the marrow specimen. The previously described fungal elements were felt to be most likely artifact, and the underlying diagnosis was reconsidered.1 This is when the repeat bone marrow examination pointed towards the diagnosis of HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a severe, systemic hyperinflammatory disorder characterized by histiocytic proliferation that may be primary or can be triggered by infection, connective tissue diseases or malignancy.25 The central pathogenesis involves dysregulated Th1 cytokine secretion. This results in an uncontrolled accumulation of activated T‐lymphocytes and histiocytes in various organs including the liver, spleen and bone marrow. The infiltration of histiocytes into major organs can lead to disruption of function and multiorgan failure.6 Viruses are the most common infectious triggers of HLH, particularly EBV, and lymphoma is the most common associated malignancy.25 It is hypothesized that EBV can interfere with normal lymphocyte signaling pathways leading to the aforementioned over‐expression of Th1 cytokines, which can then trigger HLH.7 The diagnosis of HLH is based on a combination of clinical and laboratory parameters as outlined in Table 1.8 Our patient met all five of the major criteria.
| |
| Major criteria | 1. Fever |
| 2. Splenomegaly | |
| 3. Cytopenia in two or more cell lines | |
| 4. Hypertriglyceridemia or hypofibrinogenemia | |
| 5. Hemophagocytosis on histopathologic examination | |
| Alternative criteria | A. Low or absent natural killer cell activity |
| B. Serum ferritin level >500 ug/L | |
| C. Soluble CD‐25 level >2400 U/mL | |
The recommended treatment of HLH involves the administration of the HLH‐94 protocol consisting of corticosteroids, cyclosporine and etoposide.9, 10 Those who survive this initial phase are recommended for hematopoetic stem cell transplantation producing an overall 3‐year survival rate of 64%. However, those who do not receive early etoposide therapy fare much worse, with a mortality rate of 92%.10 This patient was not able to receive etoposide because of his decompensated liver disease.
In this case, the development of Hodgkin lymphoma involving the bone marrow and liver may have resulted in a state of immune suppression. The loss of immune function likely allowed the reactivation of Epstein‐Barr virus which triggered HLH and his fulminant presentation.35,9 Indeed, both the liver and bone marrow samples showed evidence of EBV reactivation as evidenced by the presence of EBER. The diagnosis of Hodgkin lymphoma was made from a liver biopsy specimen rather than bone marrow examination. The diagnosis of Hodgkin lymphoma is based on the presence of Reed‐Sternberg cells surrounded by an inflammatory milieu of cells including variable numbers of small lymphocytes, neutrophils eosinophils and fibroblasts. The HLH‐induced pancytopenia depleted the aforementioned inflammatory milieu in the bone marrow, which obscured the diagnosis of Hodgkin lymphoma. Unfortunately, lymphoma‐associated HLH has a very poor prognosis with a mortality rate of up to 60%.4 At the outset of this case, the reported fungal elements proved to be a source of confusion which delayed the diagnosis of Hodgkin lymphoma. Given the poor prognosis of lymphoma‐associated HLH, it is unlikely this would have had any effect on the ultimate outcome.
Teaching Points
-
HLH is a rare and complex hyperinflammatory disorder which may present as pancytopenia.
-
Triggers of HLH can include infections (particularly EBV), malignancy (particularly lymphoma) and connective tissue diseases.
-
The diagnosis of HLH is based on clinical and laboratory criteria, including cytopenias that may make the evaluation for triggering conditions such as HL more difficult.
-
Lymphoma should be included in the differential diagnosis of granulomatous inflammation.
Acknowledgements
The authors acknowledge Ralph Meyer, MD (Queen's University, Kingston, Ontario) for his comments on a draft of this paper and Drs. David Barth and Maha Guindi (Department of Laboratory Medicine, University Health Network) for their reviews of the pathology specimens.
- ,Antigen detection for diagnosis of the endemic mycoses.Curr Fung Infect Rep.2008;4:189–193.
- ,,, et al.Lymphoma‐associated hemophagocytic syndrome: Clinical features and treatment outcome.Ann Hematol.2007;86:493–498.
- ,,, et al.Hodgkin lymphoma‐associated hemophagocytic syndrome: A disorder strongly correlated with Epstein‐Barr virus.Clin Inf Dis.2008;47:531–534.
- ,.Hemophagocytic syndrome associated with Hodgkin lymphoma and pneumocystis jiroveci pneumonitis.Br J Hematol.2007;138:672.
- ,,, et al.Infections associated with haemophagocytic syndrome.Lancet Infect Dis.2007;12:814–822.
- ,,,.Hemophagocytic lymphohistiocytic syndrome: Unrecognized cause of multiple organ failure.Pediatr Crit Care Med.2000;1:51–54.
- ,,, et al.Epstein‐Barr virus LMP1 Inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome.Blood.2005;106:3090–3096.
- ,,.Diagnostic guidelines for hemophagocytic lymphohistiocytosis.Semin Oncol.1991;18:29–33
- ,,, et al.Treatment of Epstein‐Barr virus‐associated hemophagocytic lymphohistiocytosis (EBV‐HLH) in young adults: A report from the HLH Study Center.Med Pediatr Oncol.2003;41:103–109.
- ,,, et al.Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis.Br J Haematol.2005;129:622–630.
- ,Antigen detection for diagnosis of the endemic mycoses.Curr Fung Infect Rep.2008;4:189–193.
- ,,, et al.Lymphoma‐associated hemophagocytic syndrome: Clinical features and treatment outcome.Ann Hematol.2007;86:493–498.
- ,,, et al.Hodgkin lymphoma‐associated hemophagocytic syndrome: A disorder strongly correlated with Epstein‐Barr virus.Clin Inf Dis.2008;47:531–534.
- ,.Hemophagocytic syndrome associated with Hodgkin lymphoma and pneumocystis jiroveci pneumonitis.Br J Hematol.2007;138:672.
- ,,, et al.Infections associated with haemophagocytic syndrome.Lancet Infect Dis.2007;12:814–822.
- ,,,.Hemophagocytic lymphohistiocytic syndrome: Unrecognized cause of multiple organ failure.Pediatr Crit Care Med.2000;1:51–54.
- ,,, et al.Epstein‐Barr virus LMP1 Inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome.Blood.2005;106:3090–3096.
- ,,.Diagnostic guidelines for hemophagocytic lymphohistiocytosis.Semin Oncol.1991;18:29–33
- ,,, et al.Treatment of Epstein‐Barr virus‐associated hemophagocytic lymphohistiocytosis (EBV‐HLH) in young adults: A report from the HLH Study Center.Med Pediatr Oncol.2003;41:103–109.
- ,,, et al.Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis.Br J Haematol.2005;129:622–630.