User login
Can’t Shake This Feeling
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
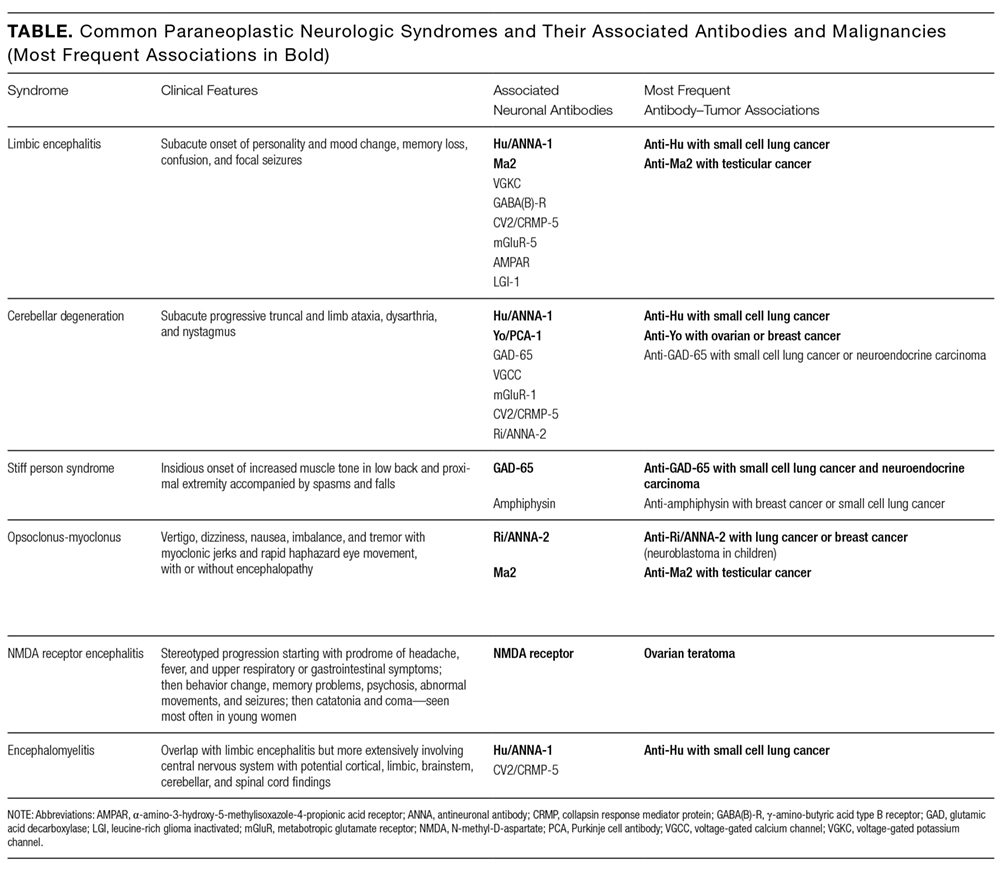
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
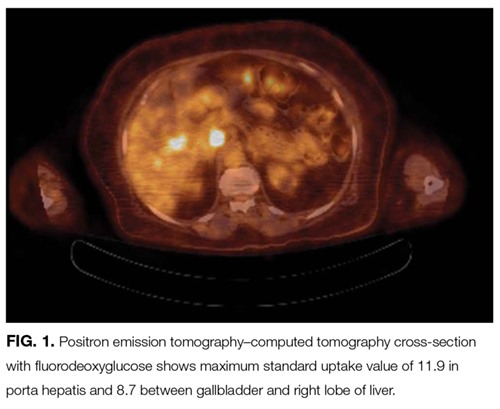
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
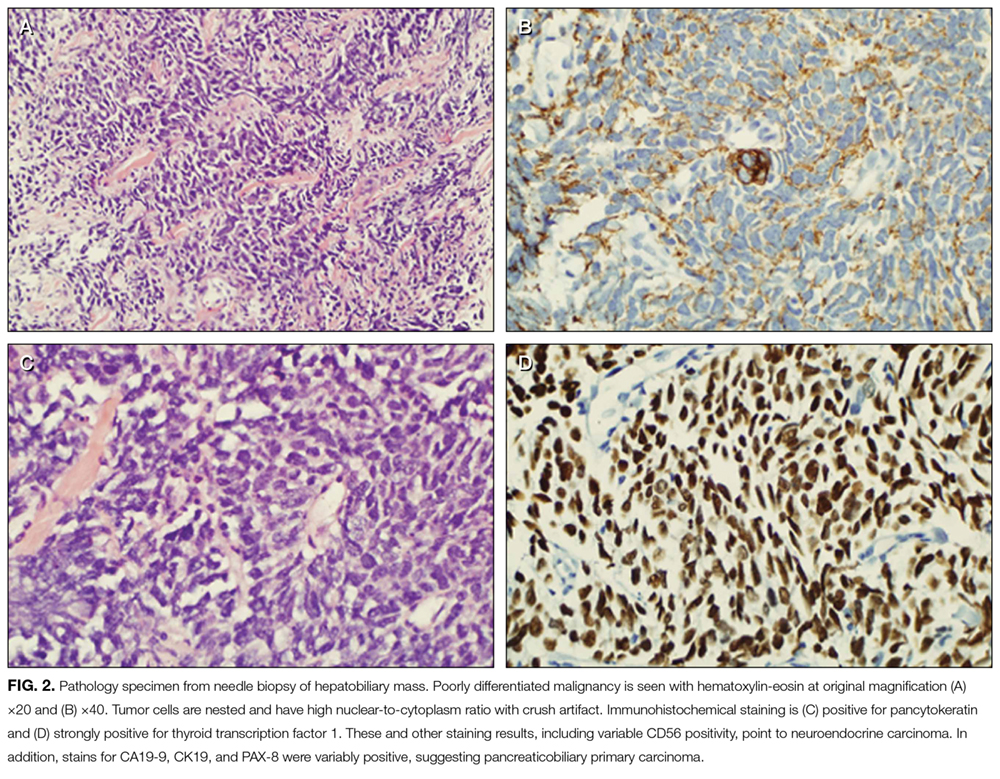
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
© 2017 Society of Hospital Medicine