User login
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA

Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12

This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency

Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
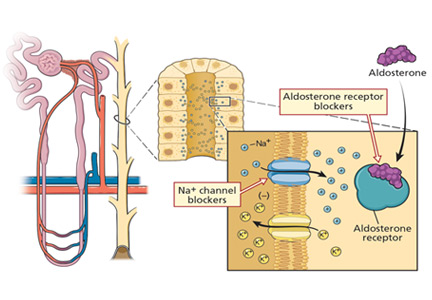
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs

Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.

Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
KEY POINTS
- Exclude pseudohyperkalemia in patients who have a normal electrocardiogram and no risk factors for the development of hyperkalemia.
- Decreased distal delivery of sodium, reduced mineralocorticoid levels or activity, and a distal tubular defect are causes of impaired renal potassium secretion.
- Medical conditions and medications that alter the renin-angiotensin-aldosterone system can give rise to hyperkalemia.