User login
Diagnosis and treatment of hyperkalemia
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA

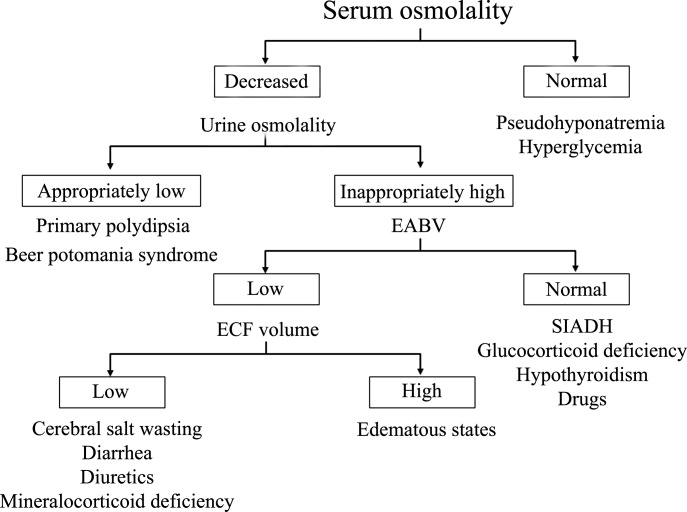
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12

This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency

Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs

Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.

Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
KEY POINTS
- Exclude pseudohyperkalemia in patients who have a normal electrocardiogram and no risk factors for the development of hyperkalemia.
- Decreased distal delivery of sodium, reduced mineralocorticoid levels or activity, and a distal tubular defect are causes of impaired renal potassium secretion.
- Medical conditions and medications that alter the renin-angiotensin-aldosterone system can give rise to hyperkalemia.
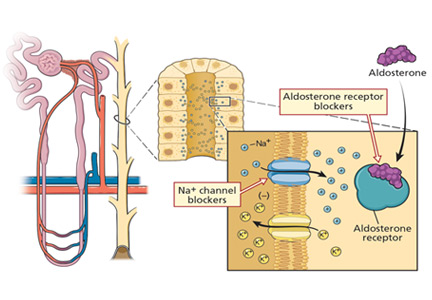
V Receptor Antagonists for Treatment of Hyponatremia
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of <10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
Conivaptan is also active as an oral formulation but its distribution has been restricted to parenteral use for short‐term (4 days maximum) in‐hospital administration only. This restriction is due to potent inhibitory effects of the drug on the hepatic cytochrome P450 3A4 enzyme system and the potential for untoward drug interactions. The inhibitory effects of other members of this class on the cytochrome P450 3A4 (CYP3A4) system are more limited, allowing for oral formulations to be used in a clinical setting.
Tolvaptan is the only other AVP antagonist currently approved by the FDA. Unlike conivaptan, tolvaptan is an oral agent with effects confined to the V2 receptor. The drug is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.
The efficacy of tolvaptan was evaluated in 2 simultaneously conducted multicenter, randomized, double blind trials called Study of Ascending Levels of Tolvaptan in hyponatremia 1 and 2 (SALT 1 and SALT 2).6 The 2 studies together randomized 225 patients with euvolemic or hypervolemic hyponatremia to outpatient treatment with the study drug for 30 days and 223 patients to placebo. Patients with a serum Na+ of <120 mEq/L in association with neurologic impairment were excluded from the trial. Serum Na+ increased more in the tolvaptan group than in the placebo during the first 4 days (3.62 vs. 0.25 mEq/L). After 30 days of therapy serum Na+ concentrations were 6.22 higher in the tolvaptan group compared with 1.66 mEq/L in the placebo group. When tolvaptan was discontinued at study end, the serum Na+ fell over a seven day period to a value similar to that in the placebo treated patients. Four patients (1.8%) exceeded the study goal of limiting correction of the hyponatremia to <12 mEq/L in the first 24 hours of treatment, but none of these patients developed adverse clinical sequelae.
As previously mentioned conivaptan and tolvaptan are indicated for the treatment of hyponatremia in the setting of euvolemia or hypervolemia. These drugs should not be used in hypovolemic states since the increase in renal water excretion can potentially predispose to worsening hemodynamics in the setting of volume depletion. To be sure, any volume of water removed from the body is principally derived from the intracellular compartment (two‐thirds) and would not be expected to affect blood pressure to a major extent. However, one‐twelfth of any water volume loss is derived from the circulating compartment and could potentially aggravate a borderline low blood pressure in the setting of volume depletion.
This concern is particularly true for conivaptan and its blocking effects on the V1a receptor. AVP can cause peripheral vasoconstriction by stimulating the V1a receptor on the peripheral vasculature.7 However, circulating concentrations observed in euvolemic and hypervolemic conditions are not typically of a magnitude to elicit this effect thus explaining the lack of clinically significant hypotension in clinical trials. By contrast, AVP may reach a high enough concentration and play a contributory role in the maintenance of blood pressure under conditions of significant depletion of extracellular fluid volume (hemorrhage) or in states of generalized vasodilation such as sepsis or advanced cirrhosis. In these settings blockade of the V1a receptor may result in significant hypotension.
Another theoretical concern of blocking the V1a receptor with conivaptan is the potential to cause further sequestration of fluid in the splanchnic vascular bed and theoretically increase the risk of hepatorenal syndrome.8 The importance of splanchnic vasodilation in the genesis of renal hypoperfusion has been indirectly illustrated by the response to ornipressin, an analog of AVP that is a preferential splanchnic vasoconstrictor. The administration of ornipressin to patients with advanced cirrhosis leads to correction of many of the systemic and renal hemodynamic abnormalities that are present. These include an elevation in mean arterial pressure, reductions in plasma renin activity and norepinephrine concentration, and increases in renal blood flow, glomerular filtration rate, and urinary sodium excretion and volume. V1a receptor blockade has the potential to increase the degree of arterial vasodilation in the splanchnic arteriolar bed. Increasing degrees of splanchnic vasodilation contribute to a fall in mean arterial pressure and unloading of baroreceptors in the central circulation. As a result, central afferent sensors signal the activation of neurohumoral effectors which in turn decrease perfusion of other organs, particularly the kidney.
AVP Antagonists in Severe Symptomatic Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, restlessness, confusion and disorientation.9 Life threatening symptoms are those of impending brain herniation and include seizures, decreased levels consciousness, and obtundation. Hypertonic saline remains the treatment of choice in those patients who are clinically determined to be severely symptomatic.10 Vaptan therapy does result in a brisk and relatively prolonged water diuresis. Indeed, in the first 6 hours following the parenteral administration of conivaptan there is a several mEq/L increase in the serum Na+ concentration.5 Whether this time course of correction is sufficient to abort fatal hyponatremic encephalopathy is simply not known. Such patients to date have been excluded from clinical studies since randomization in a placebo‐controlled trial would be clearly unethical in such subjects, and not using hypertonic saline might also be considered unsafe
Although not well studied, one could coadminister conivaptan along with hypertonic saline and anticipate a more rapid initial rise in the serum Na+. Once stabilized, the hypertonic saline could be discontinued and the remainder of correction be accomplished by use of the receptor antagonist alone. This strategy would help to minimize the likelihood of volume overload due to the use of hypertonic saline but would require frequent monitoring of the serum Na+ to ensure the rate of correction was within accepted guidelines.
AVP Antagonists in Patients With Mild to Moderate Symptoms of Hyponatremia
Hospitalized patients with mild to moderate symptoms of hyponatremia can be considered ideal candidates for the use of V2 receptor antagonists. Both conivaptan and tolvaptan can be expected to increase the serum Na+ concentration to a greater extent and more predictably than fluid restriction alone. The superiority of these drugs would be particularly evident in those who require ongoing fluid administration for any number of reasons such as parenteral administration of antibiotics or proton pump inhibitors.11
In hyponatremic patients with decompensated heart failure there may be changes in mental status in which it is difficult to separate out the contribution of hyponatremia from the decrease in cerebral perfusion. Vaptan therapy offers a predictable way to improve the serum Na+ contribution over an acceptable period of time thereby removing one of the variables. Even if hyponatremia is not the direct cause of symptoms, it may lower the threshold for mental status changes resulting from poor cerebral perfusion. A similar argument for vaptan therapy can be made in hyponatremic patients with cirrhosis. In this population mental status changes can be due to the hyponatremia, the underlying liver disease, or both. If no improvement occurred following the correction of the hyponatremia, therapy could then be focused on the underlying liver disease. In fact, in any delirious patient with hyponatremia, correction of the underlying electrolyte disorder can help to clarify the degree to which the hyponatremia is exacerbating the altered mental status.
There are no head to head studies comparing conivaptan and tolvaptan in patients with mild to moderate symptoms of hyponatremia. Presumably both drugs would work with similar efficacy. Conivaptan therapy can be complicated by phlebitis when administered through a peripheral vein due to the irritative effects of polypropylene glycol which serves as a diluent for the drug.12 For this reason the drug is often given through a central vein. The theoretical risk of conivaptan's V1a receptor blocking effects in patients with cirrhosis has already been discussed. Tolvaptan is given orally starting at 15 mg daily. Depending on the response the dose can be increased to 30 mg and ultimately to 60 mg daily. Few patients in the SALT 1 and 2 trials required the 60 mg dose.
Measurement of the urine osmolality may be useful in predicting how responsive a patient will be to the administration of a vaptan. In this regard, urine osmolality can be thought as a biomarker of the effect of AVP on the renal collecting duct.13 The higher the urine osmolality, presumably the higher the serum AVP level or the greater the effect of AVP is on the tubule. By contrast, those with a lower urine osmolality presumably have less AVP effect on the tubule and therefore will manifest a less robust response to a V2 receptor blocker.
There are circumstances unique to the hospitalized patient where one may consider use of a V2 receptor antagonist. Consider a patient admitted with pneumonia complicated by hyponatremia (Na+ = 128 mEq/L) due to syndrome of inappropriate antidiuretic hormone secretion (SIADH) who is treated with intravenous antibiotics, stabilized, and is now being considered for discharge. Despite fluid restriction the serum Na+ concentration is currently 126 mEq/L and the admitting physician is reluctant to discharge the patient for fear the Na+ may fall further and result in symptomatic hyponatremia. The hospital discharge is postponed and fluid restriction is intensified. Two days later the serum Na+ concentration has risen to 131 mEq/L and the patient is discharged. While one can question the wisdom of postponing the original planned discharge based on the lab value alone, this type of scenario is common in clinical practice. Fluid restriction is poorly tolerated, difficult to enforce, and often unpredictable in response. The administration of a single dose of a V2 receptor antagonist is a reliable way to increase the serum Na+ concentration over a 24 hour period and in this case could have potentially shortened the hospital stay.
Consider another patient admitted with a hip fracture who is found to have a serum Na+ concentration of 126 meq/L attributed to ongoing use of an antidepressant agent. The patient is otherwise medically stable but the anesthesiologist is unwilling to accept the patient for surgery until the serum Na+ is at least >130 mEq/L. Fluid restriction is prescribed and only 2 to 3 days later has the serum Na+ reached a value of 131 mEq/L. Once again, administration of a vaptan would have almost certainly increased the Na+ concentration to the desired threshold over a 24 hour period, allowing the patient to undergo the surgical procedure without experiencing the undue delay.
In each of these scenarios short term use of a vaptan would provide for a more rapid and predictable increase in the serum Na+ concentration and potentially decrease the length of hospital stay in comparison to fluid restriction alone. In addition to being less predictable and often poorly tolerated, fluid restriction is often ineffective due to the obligatory fluid administration hospitalized patients require for other therapies or nutrition. Demeclocycline and lithium have been used to antagonize the effects of AVP on the tubule but these drugs take several days before any demonstrable increase in renal water excretion is seen. Demeclocycline antagonizes the effect of AVP through nephrotoxic effects on the tubular cell whereas vaptan's are competitive inhibitors of the V2 receptor and are not associated with nephrotoxicity. Lithium interferes in the intracellular signaling pathways by which AVP causes insertion of water channels into the apical membrane. The doses required to illicit an increase in renal water excretion are near those which can result in lithium levels sufficient to cause neurotoxicity. The primary side effect of vaptan therapy are those one would predict from inducing an aquaretic effect and include thirst, increased urinary frequency, and increased urinary volume.
The decision to use a vaptan on a more prolonged basis is made on a case by case basis. When the underlying cause of increased AVP is deemed to be chronic and irreversible then therapy can be extended into the outpatient setting. Consideration for more chronic therapy would be appropriate for patients with SIADH due to underlying cancer, or those with severe chronic congestive heart failure or advanced cirrhosis. If the cause of the increased AVP is transient, then 1 to 2 doses of a vaptan while in the hospital may be all that is required. Transient causes of increased AVP would include drug related causes, acute pneumonia, hypoxia associated with respiratory failure, and acute decompensated heart failure. Once again vaptan therapy should not be used when the cause of increased AVP is due to total body salt depletion.
AVP Antagonists in Asymptomatic Hyponatremic Patients
One remaining question concerning the use of vaptan therapy is whether they are helpful in hyponatremic patients who are asymptomatic. There are several observations which raise the possibility that patients deemed to be otherwise asymptomatic do in fact have subtle abnormalities attributable to the low Na+ concentration and therefore could benefit if the hyponatremia was corrected. First, some patients with a serum Na+ in the range of 120 mEq/L to 129 mEq/L have subtle neurologic changes that improve when the serum Na+ concentration is increased.14 These include scores on tests of mental and social functioning. In the SALT 1 and 2 trials a general health survey filled out by the patients showed tolvaptan therapy was associated with improvements in the mental health component of the instrument assessing parameters such as vitality, social functioning, emotionally limited accomplishments, calmness, and sadness.6 Response time is delayed and the error rate is increased in response to various stimuli while patients are hyponatremic, suggesting hyponatremia is associated with reversible impairment in attention. It is certainly reasonable to speculate that correction of hyponatremia in an elderly patient with SIADH might improve that individual's quality of life by allowing them to better enjoy any number of activities such as reading or completing a crossword puzzle. The presence of concomitant neuropsychiatric disturbance (delirium and dementia in particular) may also be relevant, since even mild hyponatremia may increase vulnerability to further alterations in mental status in patients who are impaired at baseline.
Second, asymptomatic hyponatremic patients exhibit subtle disturbances in gait that improve following correction of the serum Na+ concentration.14, 15 In this regard, case control studies have shown an association between hyponatremia and risk of falls and fractures particularly in the elderly population.15, 16 Chronic hyponatremia has been shown to cause a reduction in bone mass in an experimental model of SIADH and hyponatremia is associated with osteoporosis in cross‐sectional human data.17 Fall‐related injury is associated with substantial adverse psychological and physical outcomes and is a cause of both death and disability in this population. It would be of considerable socioeconomic benefit if treatment of asymptomatic hyponatremia were able to reduce the risk of this complication.
Lastly, hyponatremia is associated with increased morbidity and mortality but generally the low Na+ concentration is merely thought to be marker of the severity of the underlying disease and not a direct contributor to the adverse outcome.18, 19 For example in decompensated heart failure there is an inverse relationship between the degree of hyponatremia and the extent of neurohormonal activation. Adverse effects resulting from persistent activation of the renin‐angiotensin‐aldosterone system and sympathetic nerve activity are thought to be the mechanism underlying the increase in mortality in hyponatremic patients with heart failure. Nevertheless, it has been suggested that hyponatremia itself, the associated hypotonicity, and/or elevated levels of AVP might exert adverse effects on the cardiovascular system or other organ systems and therefore play a contributory role in patient morbidity and mortality. While it remains speculative as to whether correction of the hyponatremia per se will improve patient outcomes, the V2 receptor antagonists offer an opportunity to test this uncertainty in patients with euvolemic and hypervolemic hyponatremia.0, 0
| Subtype | Location | Function |
|---|---|---|
| ||
| V1A | Vascular smooth muscle to include splanchnic bed | Vasoconstriction |
| V1B | Anterior pituitary | Release of ACTH |
| V2 | Basolateral surface of renal collecting duct, vascular endothelium, vascular smooth muscle | Insertion of aquaporin channel into luminal membrane, release of von Willebrand factor, vasodilation |
| Parameter | Tolvaptan | Conivaptan |
|---|---|---|
| ||
| Trade name | Samsca | Vaprisol |
| Administration | Oral | Intravenous |
| Dose | 1560 mg daily | 20 mg loading dose followed by 2040 mg continuous infusion |
| Receptor | V2 | V1A and V2 |
| Protein binding (%) | 99 | 98.5 |
| Half life | 68 hours | 38 hours |
| Metabolism | Hepatic (CYP3A4) | Hepatic (CYP3A4) |
| Elimination | Feces | Feces |
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of <10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
Conivaptan is also active as an oral formulation but its distribution has been restricted to parenteral use for short‐term (4 days maximum) in‐hospital administration only. This restriction is due to potent inhibitory effects of the drug on the hepatic cytochrome P450 3A4 enzyme system and the potential for untoward drug interactions. The inhibitory effects of other members of this class on the cytochrome P450 3A4 (CYP3A4) system are more limited, allowing for oral formulations to be used in a clinical setting.
Tolvaptan is the only other AVP antagonist currently approved by the FDA. Unlike conivaptan, tolvaptan is an oral agent with effects confined to the V2 receptor. The drug is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.
The efficacy of tolvaptan was evaluated in 2 simultaneously conducted multicenter, randomized, double blind trials called Study of Ascending Levels of Tolvaptan in hyponatremia 1 and 2 (SALT 1 and SALT 2).6 The 2 studies together randomized 225 patients with euvolemic or hypervolemic hyponatremia to outpatient treatment with the study drug for 30 days and 223 patients to placebo. Patients with a serum Na+ of <120 mEq/L in association with neurologic impairment were excluded from the trial. Serum Na+ increased more in the tolvaptan group than in the placebo during the first 4 days (3.62 vs. 0.25 mEq/L). After 30 days of therapy serum Na+ concentrations were 6.22 higher in the tolvaptan group compared with 1.66 mEq/L in the placebo group. When tolvaptan was discontinued at study end, the serum Na+ fell over a seven day period to a value similar to that in the placebo treated patients. Four patients (1.8%) exceeded the study goal of limiting correction of the hyponatremia to <12 mEq/L in the first 24 hours of treatment, but none of these patients developed adverse clinical sequelae.
As previously mentioned conivaptan and tolvaptan are indicated for the treatment of hyponatremia in the setting of euvolemia or hypervolemia. These drugs should not be used in hypovolemic states since the increase in renal water excretion can potentially predispose to worsening hemodynamics in the setting of volume depletion. To be sure, any volume of water removed from the body is principally derived from the intracellular compartment (two‐thirds) and would not be expected to affect blood pressure to a major extent. However, one‐twelfth of any water volume loss is derived from the circulating compartment and could potentially aggravate a borderline low blood pressure in the setting of volume depletion.
This concern is particularly true for conivaptan and its blocking effects on the V1a receptor. AVP can cause peripheral vasoconstriction by stimulating the V1a receptor on the peripheral vasculature.7 However, circulating concentrations observed in euvolemic and hypervolemic conditions are not typically of a magnitude to elicit this effect thus explaining the lack of clinically significant hypotension in clinical trials. By contrast, AVP may reach a high enough concentration and play a contributory role in the maintenance of blood pressure under conditions of significant depletion of extracellular fluid volume (hemorrhage) or in states of generalized vasodilation such as sepsis or advanced cirrhosis. In these settings blockade of the V1a receptor may result in significant hypotension.
Another theoretical concern of blocking the V1a receptor with conivaptan is the potential to cause further sequestration of fluid in the splanchnic vascular bed and theoretically increase the risk of hepatorenal syndrome.8 The importance of splanchnic vasodilation in the genesis of renal hypoperfusion has been indirectly illustrated by the response to ornipressin, an analog of AVP that is a preferential splanchnic vasoconstrictor. The administration of ornipressin to patients with advanced cirrhosis leads to correction of many of the systemic and renal hemodynamic abnormalities that are present. These include an elevation in mean arterial pressure, reductions in plasma renin activity and norepinephrine concentration, and increases in renal blood flow, glomerular filtration rate, and urinary sodium excretion and volume. V1a receptor blockade has the potential to increase the degree of arterial vasodilation in the splanchnic arteriolar bed. Increasing degrees of splanchnic vasodilation contribute to a fall in mean arterial pressure and unloading of baroreceptors in the central circulation. As a result, central afferent sensors signal the activation of neurohumoral effectors which in turn decrease perfusion of other organs, particularly the kidney.
AVP Antagonists in Severe Symptomatic Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, restlessness, confusion and disorientation.9 Life threatening symptoms are those of impending brain herniation and include seizures, decreased levels consciousness, and obtundation. Hypertonic saline remains the treatment of choice in those patients who are clinically determined to be severely symptomatic.10 Vaptan therapy does result in a brisk and relatively prolonged water diuresis. Indeed, in the first 6 hours following the parenteral administration of conivaptan there is a several mEq/L increase in the serum Na+ concentration.5 Whether this time course of correction is sufficient to abort fatal hyponatremic encephalopathy is simply not known. Such patients to date have been excluded from clinical studies since randomization in a placebo‐controlled trial would be clearly unethical in such subjects, and not using hypertonic saline might also be considered unsafe
Although not well studied, one could coadminister conivaptan along with hypertonic saline and anticipate a more rapid initial rise in the serum Na+. Once stabilized, the hypertonic saline could be discontinued and the remainder of correction be accomplished by use of the receptor antagonist alone. This strategy would help to minimize the likelihood of volume overload due to the use of hypertonic saline but would require frequent monitoring of the serum Na+ to ensure the rate of correction was within accepted guidelines.
AVP Antagonists in Patients With Mild to Moderate Symptoms of Hyponatremia
Hospitalized patients with mild to moderate symptoms of hyponatremia can be considered ideal candidates for the use of V2 receptor antagonists. Both conivaptan and tolvaptan can be expected to increase the serum Na+ concentration to a greater extent and more predictably than fluid restriction alone. The superiority of these drugs would be particularly evident in those who require ongoing fluid administration for any number of reasons such as parenteral administration of antibiotics or proton pump inhibitors.11
In hyponatremic patients with decompensated heart failure there may be changes in mental status in which it is difficult to separate out the contribution of hyponatremia from the decrease in cerebral perfusion. Vaptan therapy offers a predictable way to improve the serum Na+ contribution over an acceptable period of time thereby removing one of the variables. Even if hyponatremia is not the direct cause of symptoms, it may lower the threshold for mental status changes resulting from poor cerebral perfusion. A similar argument for vaptan therapy can be made in hyponatremic patients with cirrhosis. In this population mental status changes can be due to the hyponatremia, the underlying liver disease, or both. If no improvement occurred following the correction of the hyponatremia, therapy could then be focused on the underlying liver disease. In fact, in any delirious patient with hyponatremia, correction of the underlying electrolyte disorder can help to clarify the degree to which the hyponatremia is exacerbating the altered mental status.
There are no head to head studies comparing conivaptan and tolvaptan in patients with mild to moderate symptoms of hyponatremia. Presumably both drugs would work with similar efficacy. Conivaptan therapy can be complicated by phlebitis when administered through a peripheral vein due to the irritative effects of polypropylene glycol which serves as a diluent for the drug.12 For this reason the drug is often given through a central vein. The theoretical risk of conivaptan's V1a receptor blocking effects in patients with cirrhosis has already been discussed. Tolvaptan is given orally starting at 15 mg daily. Depending on the response the dose can be increased to 30 mg and ultimately to 60 mg daily. Few patients in the SALT 1 and 2 trials required the 60 mg dose.
Measurement of the urine osmolality may be useful in predicting how responsive a patient will be to the administration of a vaptan. In this regard, urine osmolality can be thought as a biomarker of the effect of AVP on the renal collecting duct.13 The higher the urine osmolality, presumably the higher the serum AVP level or the greater the effect of AVP is on the tubule. By contrast, those with a lower urine osmolality presumably have less AVP effect on the tubule and therefore will manifest a less robust response to a V2 receptor blocker.
There are circumstances unique to the hospitalized patient where one may consider use of a V2 receptor antagonist. Consider a patient admitted with pneumonia complicated by hyponatremia (Na+ = 128 mEq/L) due to syndrome of inappropriate antidiuretic hormone secretion (SIADH) who is treated with intravenous antibiotics, stabilized, and is now being considered for discharge. Despite fluid restriction the serum Na+ concentration is currently 126 mEq/L and the admitting physician is reluctant to discharge the patient for fear the Na+ may fall further and result in symptomatic hyponatremia. The hospital discharge is postponed and fluid restriction is intensified. Two days later the serum Na+ concentration has risen to 131 mEq/L and the patient is discharged. While one can question the wisdom of postponing the original planned discharge based on the lab value alone, this type of scenario is common in clinical practice. Fluid restriction is poorly tolerated, difficult to enforce, and often unpredictable in response. The administration of a single dose of a V2 receptor antagonist is a reliable way to increase the serum Na+ concentration over a 24 hour period and in this case could have potentially shortened the hospital stay.
Consider another patient admitted with a hip fracture who is found to have a serum Na+ concentration of 126 meq/L attributed to ongoing use of an antidepressant agent. The patient is otherwise medically stable but the anesthesiologist is unwilling to accept the patient for surgery until the serum Na+ is at least >130 mEq/L. Fluid restriction is prescribed and only 2 to 3 days later has the serum Na+ reached a value of 131 mEq/L. Once again, administration of a vaptan would have almost certainly increased the Na+ concentration to the desired threshold over a 24 hour period, allowing the patient to undergo the surgical procedure without experiencing the undue delay.
In each of these scenarios short term use of a vaptan would provide for a more rapid and predictable increase in the serum Na+ concentration and potentially decrease the length of hospital stay in comparison to fluid restriction alone. In addition to being less predictable and often poorly tolerated, fluid restriction is often ineffective due to the obligatory fluid administration hospitalized patients require for other therapies or nutrition. Demeclocycline and lithium have been used to antagonize the effects of AVP on the tubule but these drugs take several days before any demonstrable increase in renal water excretion is seen. Demeclocycline antagonizes the effect of AVP through nephrotoxic effects on the tubular cell whereas vaptan's are competitive inhibitors of the V2 receptor and are not associated with nephrotoxicity. Lithium interferes in the intracellular signaling pathways by which AVP causes insertion of water channels into the apical membrane. The doses required to illicit an increase in renal water excretion are near those which can result in lithium levels sufficient to cause neurotoxicity. The primary side effect of vaptan therapy are those one would predict from inducing an aquaretic effect and include thirst, increased urinary frequency, and increased urinary volume.
The decision to use a vaptan on a more prolonged basis is made on a case by case basis. When the underlying cause of increased AVP is deemed to be chronic and irreversible then therapy can be extended into the outpatient setting. Consideration for more chronic therapy would be appropriate for patients with SIADH due to underlying cancer, or those with severe chronic congestive heart failure or advanced cirrhosis. If the cause of the increased AVP is transient, then 1 to 2 doses of a vaptan while in the hospital may be all that is required. Transient causes of increased AVP would include drug related causes, acute pneumonia, hypoxia associated with respiratory failure, and acute decompensated heart failure. Once again vaptan therapy should not be used when the cause of increased AVP is due to total body salt depletion.
AVP Antagonists in Asymptomatic Hyponatremic Patients
One remaining question concerning the use of vaptan therapy is whether they are helpful in hyponatremic patients who are asymptomatic. There are several observations which raise the possibility that patients deemed to be otherwise asymptomatic do in fact have subtle abnormalities attributable to the low Na+ concentration and therefore could benefit if the hyponatremia was corrected. First, some patients with a serum Na+ in the range of 120 mEq/L to 129 mEq/L have subtle neurologic changes that improve when the serum Na+ concentration is increased.14 These include scores on tests of mental and social functioning. In the SALT 1 and 2 trials a general health survey filled out by the patients showed tolvaptan therapy was associated with improvements in the mental health component of the instrument assessing parameters such as vitality, social functioning, emotionally limited accomplishments, calmness, and sadness.6 Response time is delayed and the error rate is increased in response to various stimuli while patients are hyponatremic, suggesting hyponatremia is associated with reversible impairment in attention. It is certainly reasonable to speculate that correction of hyponatremia in an elderly patient with SIADH might improve that individual's quality of life by allowing them to better enjoy any number of activities such as reading or completing a crossword puzzle. The presence of concomitant neuropsychiatric disturbance (delirium and dementia in particular) may also be relevant, since even mild hyponatremia may increase vulnerability to further alterations in mental status in patients who are impaired at baseline.
Second, asymptomatic hyponatremic patients exhibit subtle disturbances in gait that improve following correction of the serum Na+ concentration.14, 15 In this regard, case control studies have shown an association between hyponatremia and risk of falls and fractures particularly in the elderly population.15, 16 Chronic hyponatremia has been shown to cause a reduction in bone mass in an experimental model of SIADH and hyponatremia is associated with osteoporosis in cross‐sectional human data.17 Fall‐related injury is associated with substantial adverse psychological and physical outcomes and is a cause of both death and disability in this population. It would be of considerable socioeconomic benefit if treatment of asymptomatic hyponatremia were able to reduce the risk of this complication.
Lastly, hyponatremia is associated with increased morbidity and mortality but generally the low Na+ concentration is merely thought to be marker of the severity of the underlying disease and not a direct contributor to the adverse outcome.18, 19 For example in decompensated heart failure there is an inverse relationship between the degree of hyponatremia and the extent of neurohormonal activation. Adverse effects resulting from persistent activation of the renin‐angiotensin‐aldosterone system and sympathetic nerve activity are thought to be the mechanism underlying the increase in mortality in hyponatremic patients with heart failure. Nevertheless, it has been suggested that hyponatremia itself, the associated hypotonicity, and/or elevated levels of AVP might exert adverse effects on the cardiovascular system or other organ systems and therefore play a contributory role in patient morbidity and mortality. While it remains speculative as to whether correction of the hyponatremia per se will improve patient outcomes, the V2 receptor antagonists offer an opportunity to test this uncertainty in patients with euvolemic and hypervolemic hyponatremia.0, 0
| Subtype | Location | Function |
|---|---|---|
| ||
| V1A | Vascular smooth muscle to include splanchnic bed | Vasoconstriction |
| V1B | Anterior pituitary | Release of ACTH |
| V2 | Basolateral surface of renal collecting duct, vascular endothelium, vascular smooth muscle | Insertion of aquaporin channel into luminal membrane, release of von Willebrand factor, vasodilation |
| Parameter | Tolvaptan | Conivaptan |
|---|---|---|
| ||
| Trade name | Samsca | Vaprisol |
| Administration | Oral | Intravenous |
| Dose | 1560 mg daily | 20 mg loading dose followed by 2040 mg continuous infusion |
| Receptor | V2 | V1A and V2 |
| Protein binding (%) | 99 | 98.5 |
| Half life | 68 hours | 38 hours |
| Metabolism | Hepatic (CYP3A4) | Hepatic (CYP3A4) |
| Elimination | Feces | Feces |
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of <10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
Conivaptan is also active as an oral formulation but its distribution has been restricted to parenteral use for short‐term (4 days maximum) in‐hospital administration only. This restriction is due to potent inhibitory effects of the drug on the hepatic cytochrome P450 3A4 enzyme system and the potential for untoward drug interactions. The inhibitory effects of other members of this class on the cytochrome P450 3A4 (CYP3A4) system are more limited, allowing for oral formulations to be used in a clinical setting.
Tolvaptan is the only other AVP antagonist currently approved by the FDA. Unlike conivaptan, tolvaptan is an oral agent with effects confined to the V2 receptor. The drug is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.
The efficacy of tolvaptan was evaluated in 2 simultaneously conducted multicenter, randomized, double blind trials called Study of Ascending Levels of Tolvaptan in hyponatremia 1 and 2 (SALT 1 and SALT 2).6 The 2 studies together randomized 225 patients with euvolemic or hypervolemic hyponatremia to outpatient treatment with the study drug for 30 days and 223 patients to placebo. Patients with a serum Na+ of <120 mEq/L in association with neurologic impairment were excluded from the trial. Serum Na+ increased more in the tolvaptan group than in the placebo during the first 4 days (3.62 vs. 0.25 mEq/L). After 30 days of therapy serum Na+ concentrations were 6.22 higher in the tolvaptan group compared with 1.66 mEq/L in the placebo group. When tolvaptan was discontinued at study end, the serum Na+ fell over a seven day period to a value similar to that in the placebo treated patients. Four patients (1.8%) exceeded the study goal of limiting correction of the hyponatremia to <12 mEq/L in the first 24 hours of treatment, but none of these patients developed adverse clinical sequelae.
As previously mentioned conivaptan and tolvaptan are indicated for the treatment of hyponatremia in the setting of euvolemia or hypervolemia. These drugs should not be used in hypovolemic states since the increase in renal water excretion can potentially predispose to worsening hemodynamics in the setting of volume depletion. To be sure, any volume of water removed from the body is principally derived from the intracellular compartment (two‐thirds) and would not be expected to affect blood pressure to a major extent. However, one‐twelfth of any water volume loss is derived from the circulating compartment and could potentially aggravate a borderline low blood pressure in the setting of volume depletion.
This concern is particularly true for conivaptan and its blocking effects on the V1a receptor. AVP can cause peripheral vasoconstriction by stimulating the V1a receptor on the peripheral vasculature.7 However, circulating concentrations observed in euvolemic and hypervolemic conditions are not typically of a magnitude to elicit this effect thus explaining the lack of clinically significant hypotension in clinical trials. By contrast, AVP may reach a high enough concentration and play a contributory role in the maintenance of blood pressure under conditions of significant depletion of extracellular fluid volume (hemorrhage) or in states of generalized vasodilation such as sepsis or advanced cirrhosis. In these settings blockade of the V1a receptor may result in significant hypotension.
Another theoretical concern of blocking the V1a receptor with conivaptan is the potential to cause further sequestration of fluid in the splanchnic vascular bed and theoretically increase the risk of hepatorenal syndrome.8 The importance of splanchnic vasodilation in the genesis of renal hypoperfusion has been indirectly illustrated by the response to ornipressin, an analog of AVP that is a preferential splanchnic vasoconstrictor. The administration of ornipressin to patients with advanced cirrhosis leads to correction of many of the systemic and renal hemodynamic abnormalities that are present. These include an elevation in mean arterial pressure, reductions in plasma renin activity and norepinephrine concentration, and increases in renal blood flow, glomerular filtration rate, and urinary sodium excretion and volume. V1a receptor blockade has the potential to increase the degree of arterial vasodilation in the splanchnic arteriolar bed. Increasing degrees of splanchnic vasodilation contribute to a fall in mean arterial pressure and unloading of baroreceptors in the central circulation. As a result, central afferent sensors signal the activation of neurohumoral effectors which in turn decrease perfusion of other organs, particularly the kidney.
AVP Antagonists in Severe Symptomatic Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, restlessness, confusion and disorientation.9 Life threatening symptoms are those of impending brain herniation and include seizures, decreased levels consciousness, and obtundation. Hypertonic saline remains the treatment of choice in those patients who are clinically determined to be severely symptomatic.10 Vaptan therapy does result in a brisk and relatively prolonged water diuresis. Indeed, in the first 6 hours following the parenteral administration of conivaptan there is a several mEq/L increase in the serum Na+ concentration.5 Whether this time course of correction is sufficient to abort fatal hyponatremic encephalopathy is simply not known. Such patients to date have been excluded from clinical studies since randomization in a placebo‐controlled trial would be clearly unethical in such subjects, and not using hypertonic saline might also be considered unsafe
Although not well studied, one could coadminister conivaptan along with hypertonic saline and anticipate a more rapid initial rise in the serum Na+. Once stabilized, the hypertonic saline could be discontinued and the remainder of correction be accomplished by use of the receptor antagonist alone. This strategy would help to minimize the likelihood of volume overload due to the use of hypertonic saline but would require frequent monitoring of the serum Na+ to ensure the rate of correction was within accepted guidelines.
AVP Antagonists in Patients With Mild to Moderate Symptoms of Hyponatremia
Hospitalized patients with mild to moderate symptoms of hyponatremia can be considered ideal candidates for the use of V2 receptor antagonists. Both conivaptan and tolvaptan can be expected to increase the serum Na+ concentration to a greater extent and more predictably than fluid restriction alone. The superiority of these drugs would be particularly evident in those who require ongoing fluid administration for any number of reasons such as parenteral administration of antibiotics or proton pump inhibitors.11
In hyponatremic patients with decompensated heart failure there may be changes in mental status in which it is difficult to separate out the contribution of hyponatremia from the decrease in cerebral perfusion. Vaptan therapy offers a predictable way to improve the serum Na+ contribution over an acceptable period of time thereby removing one of the variables. Even if hyponatremia is not the direct cause of symptoms, it may lower the threshold for mental status changes resulting from poor cerebral perfusion. A similar argument for vaptan therapy can be made in hyponatremic patients with cirrhosis. In this population mental status changes can be due to the hyponatremia, the underlying liver disease, or both. If no improvement occurred following the correction of the hyponatremia, therapy could then be focused on the underlying liver disease. In fact, in any delirious patient with hyponatremia, correction of the underlying electrolyte disorder can help to clarify the degree to which the hyponatremia is exacerbating the altered mental status.
There are no head to head studies comparing conivaptan and tolvaptan in patients with mild to moderate symptoms of hyponatremia. Presumably both drugs would work with similar efficacy. Conivaptan therapy can be complicated by phlebitis when administered through a peripheral vein due to the irritative effects of polypropylene glycol which serves as a diluent for the drug.12 For this reason the drug is often given through a central vein. The theoretical risk of conivaptan's V1a receptor blocking effects in patients with cirrhosis has already been discussed. Tolvaptan is given orally starting at 15 mg daily. Depending on the response the dose can be increased to 30 mg and ultimately to 60 mg daily. Few patients in the SALT 1 and 2 trials required the 60 mg dose.
Measurement of the urine osmolality may be useful in predicting how responsive a patient will be to the administration of a vaptan. In this regard, urine osmolality can be thought as a biomarker of the effect of AVP on the renal collecting duct.13 The higher the urine osmolality, presumably the higher the serum AVP level or the greater the effect of AVP is on the tubule. By contrast, those with a lower urine osmolality presumably have less AVP effect on the tubule and therefore will manifest a less robust response to a V2 receptor blocker.
There are circumstances unique to the hospitalized patient where one may consider use of a V2 receptor antagonist. Consider a patient admitted with pneumonia complicated by hyponatremia (Na+ = 128 mEq/L) due to syndrome of inappropriate antidiuretic hormone secretion (SIADH) who is treated with intravenous antibiotics, stabilized, and is now being considered for discharge. Despite fluid restriction the serum Na+ concentration is currently 126 mEq/L and the admitting physician is reluctant to discharge the patient for fear the Na+ may fall further and result in symptomatic hyponatremia. The hospital discharge is postponed and fluid restriction is intensified. Two days later the serum Na+ concentration has risen to 131 mEq/L and the patient is discharged. While one can question the wisdom of postponing the original planned discharge based on the lab value alone, this type of scenario is common in clinical practice. Fluid restriction is poorly tolerated, difficult to enforce, and often unpredictable in response. The administration of a single dose of a V2 receptor antagonist is a reliable way to increase the serum Na+ concentration over a 24 hour period and in this case could have potentially shortened the hospital stay.
Consider another patient admitted with a hip fracture who is found to have a serum Na+ concentration of 126 meq/L attributed to ongoing use of an antidepressant agent. The patient is otherwise medically stable but the anesthesiologist is unwilling to accept the patient for surgery until the serum Na+ is at least >130 mEq/L. Fluid restriction is prescribed and only 2 to 3 days later has the serum Na+ reached a value of 131 mEq/L. Once again, administration of a vaptan would have almost certainly increased the Na+ concentration to the desired threshold over a 24 hour period, allowing the patient to undergo the surgical procedure without experiencing the undue delay.
In each of these scenarios short term use of a vaptan would provide for a more rapid and predictable increase in the serum Na+ concentration and potentially decrease the length of hospital stay in comparison to fluid restriction alone. In addition to being less predictable and often poorly tolerated, fluid restriction is often ineffective due to the obligatory fluid administration hospitalized patients require for other therapies or nutrition. Demeclocycline and lithium have been used to antagonize the effects of AVP on the tubule but these drugs take several days before any demonstrable increase in renal water excretion is seen. Demeclocycline antagonizes the effect of AVP through nephrotoxic effects on the tubular cell whereas vaptan's are competitive inhibitors of the V2 receptor and are not associated with nephrotoxicity. Lithium interferes in the intracellular signaling pathways by which AVP causes insertion of water channels into the apical membrane. The doses required to illicit an increase in renal water excretion are near those which can result in lithium levels sufficient to cause neurotoxicity. The primary side effect of vaptan therapy are those one would predict from inducing an aquaretic effect and include thirst, increased urinary frequency, and increased urinary volume.
The decision to use a vaptan on a more prolonged basis is made on a case by case basis. When the underlying cause of increased AVP is deemed to be chronic and irreversible then therapy can be extended into the outpatient setting. Consideration for more chronic therapy would be appropriate for patients with SIADH due to underlying cancer, or those with severe chronic congestive heart failure or advanced cirrhosis. If the cause of the increased AVP is transient, then 1 to 2 doses of a vaptan while in the hospital may be all that is required. Transient causes of increased AVP would include drug related causes, acute pneumonia, hypoxia associated with respiratory failure, and acute decompensated heart failure. Once again vaptan therapy should not be used when the cause of increased AVP is due to total body salt depletion.
AVP Antagonists in Asymptomatic Hyponatremic Patients
One remaining question concerning the use of vaptan therapy is whether they are helpful in hyponatremic patients who are asymptomatic. There are several observations which raise the possibility that patients deemed to be otherwise asymptomatic do in fact have subtle abnormalities attributable to the low Na+ concentration and therefore could benefit if the hyponatremia was corrected. First, some patients with a serum Na+ in the range of 120 mEq/L to 129 mEq/L have subtle neurologic changes that improve when the serum Na+ concentration is increased.14 These include scores on tests of mental and social functioning. In the SALT 1 and 2 trials a general health survey filled out by the patients showed tolvaptan therapy was associated with improvements in the mental health component of the instrument assessing parameters such as vitality, social functioning, emotionally limited accomplishments, calmness, and sadness.6 Response time is delayed and the error rate is increased in response to various stimuli while patients are hyponatremic, suggesting hyponatremia is associated with reversible impairment in attention. It is certainly reasonable to speculate that correction of hyponatremia in an elderly patient with SIADH might improve that individual's quality of life by allowing them to better enjoy any number of activities such as reading or completing a crossword puzzle. The presence of concomitant neuropsychiatric disturbance (delirium and dementia in particular) may also be relevant, since even mild hyponatremia may increase vulnerability to further alterations in mental status in patients who are impaired at baseline.
Second, asymptomatic hyponatremic patients exhibit subtle disturbances in gait that improve following correction of the serum Na+ concentration.14, 15 In this regard, case control studies have shown an association between hyponatremia and risk of falls and fractures particularly in the elderly population.15, 16 Chronic hyponatremia has been shown to cause a reduction in bone mass in an experimental model of SIADH and hyponatremia is associated with osteoporosis in cross‐sectional human data.17 Fall‐related injury is associated with substantial adverse psychological and physical outcomes and is a cause of both death and disability in this population. It would be of considerable socioeconomic benefit if treatment of asymptomatic hyponatremia were able to reduce the risk of this complication.
Lastly, hyponatremia is associated with increased morbidity and mortality but generally the low Na+ concentration is merely thought to be marker of the severity of the underlying disease and not a direct contributor to the adverse outcome.18, 19 For example in decompensated heart failure there is an inverse relationship between the degree of hyponatremia and the extent of neurohormonal activation. Adverse effects resulting from persistent activation of the renin‐angiotensin‐aldosterone system and sympathetic nerve activity are thought to be the mechanism underlying the increase in mortality in hyponatremic patients with heart failure. Nevertheless, it has been suggested that hyponatremia itself, the associated hypotonicity, and/or elevated levels of AVP might exert adverse effects on the cardiovascular system or other organ systems and therefore play a contributory role in patient morbidity and mortality. While it remains speculative as to whether correction of the hyponatremia per se will improve patient outcomes, the V2 receptor antagonists offer an opportunity to test this uncertainty in patients with euvolemic and hypervolemic hyponatremia.0, 0
| Subtype | Location | Function |
|---|---|---|
| ||
| V1A | Vascular smooth muscle to include splanchnic bed | Vasoconstriction |
| V1B | Anterior pituitary | Release of ACTH |
| V2 | Basolateral surface of renal collecting duct, vascular endothelium, vascular smooth muscle | Insertion of aquaporin channel into luminal membrane, release of von Willebrand factor, vasodilation |
| Parameter | Tolvaptan | Conivaptan |
|---|---|---|
| ||
| Trade name | Samsca | Vaprisol |
| Administration | Oral | Intravenous |
| Dose | 1560 mg daily | 20 mg loading dose followed by 2040 mg continuous infusion |
| Receptor | V2 | V1A and V2 |
| Protein binding (%) | 99 | 98.5 |
| Half life | 68 hours | 38 hours |
| Metabolism | Hepatic (CYP3A4) | Hepatic (CYP3A4) |
| Elimination | Feces | Feces |
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
Clinical Approach and Treatment of the Hyponatremic Patient
Hyponatremia is one of the most common electrolyte abnormalities encountered in clinical practice. The frequency of the disorder varies according to definition and clinical setting but has been reported to be present in 28% of patients upon hospital admission and in 7% of patients attending an ambulatory community clinic.1 Increasing age, medications, various disease states, and administration of hypotonic fluids are among the known risk factors for the disorder.
The mortality rate in hyponatremic patients is approximately 3 times that of normonatremic hospitalized patients.25 Outcomes are particularly poor in those patients whose serum sodium (Na+) falls during a hospitalization. In 1 prospective study the mortality rate in patients with a normal serum Na+ concentration was 0.2% in comparison to a mortality rate of 11.2% and 25% in patients with a serum Na+ concentration <130 mEq/L and <120 mEq/L, respectively.2 In a recent retrospective cohort study of 10,899 hospitalized patients, the incidence of hyponatremia (<135 mmol/L) at admission was 5.5%.5 As compared to those with normonatremia, these patients were more likely to require intensive care and mechanical ventilation within 48 hours of hospitalization. In addition, hospital mortality, mean length of stay, and costs were significantly greater among patients with hyponatremia than those without.
The association with hyponatremia and adverse outcomes could be the direct result of hyponatremia, the comormidities that lead to the electrolyte derangement, or both. Whatever the mechanism, hyponatremia should not be viewed as an innocuous condition. Rather, clinicians should view this disorder with urgency and institute measures to prevent any further decline in the serum Na+ concentration and initiate appropriate therapy for its correction. This review will first briefly summarize the pathogenesis of hyponatremia and then discuss various disease states encountered in the hospital setting in which hyponatremia is frequently present.
Pathogenesis of Hyponatremia
Hyponatremia is generally associated with a hypoosmolar state and is a marker for a disturbance in water balance. Stated differently, all hyponatremia is dilutional. The approach to the patient with hyponatremia is outlined in Figure 1.
Is the Hyponatremia Representative of a Hypoosmolar State?
There are 3 causes of hyponatremia in which it is not associated with a hypoosmolar state. The first of these is pseudohyponatremia which involves an abnormal measurement of the serum Na+. This occurs in patients with hyperglobulinemia or hypertriglyceridemia in whom plasma water relative to plasma solids is decreased in blood, leading to less Na+ in a given volume of blood. In general, this problem is becoming less prevalent as many laboratories are using Na+ electrodes without diluting the blood such that the Na+ measurement becomes independent of plasma water and nonaqueous contents.
A second cause of hyponatremia in the absence of a hypoosmolar state involves true hyponatremia but with elevations in the concentration of another osmole. Clinical examples include hyperglycemia as seen in uncontrolled diabetes or rarely hypertonic infusion of mannitol used in the treatment of cerebral edema. The accumulation of these effective osmoles creates an osmotic force causing water to move from the intracellular to the extracellular space thus diluting the serum Na+. For every 100 mg/dL rise in glucose or mannitol, the serum Na+ will quickly fall by 1.6 mEq/L. This increase in tonicity also stimulates thirst and arginine vasopressin (AVP) secretion, both of which contribute to further water retention. As a result the plasma osmolality and serum Na+ concentration will continue to fall. Once the plasma osmolality normalizes the serum Na+ will have decreased by 2.8 mEq/L for every 100 mg/dL rise in glucose.
The third cause of hyponatremia in the absence of a hypoosmolar state is the addition of an isosmotic (or near isosmotic) non‐Na+ containing fluid to the extracellular space. This situation typically occurs during a transurethral resection of the prostate or during laprascopic surgery when large amounts of a nonconducting flushing solution containing glycine or sorbitol are absorbed systemically.
Is the Kidney's Ability to Dilute the Urine Intact?
The presence of hypotonic hyponatremia implies that water intake exceeds the ability of the kidney to excrete water. In unusual circumstances, this can occur when the kidneys ability to excrete free water is intact. However, because a normal kidney can excrete 18 L of water per day, the presence of hyponatremia with normal renal water excretion implies the patient is drinking >20 L water/day. This condition is referred to as primary polydipsia. These patients should have a urine osmolality <100 mOsm/L. While primary polydipsia is a common condition which leads to polyuria and polydipsia, it is uncommon as a sole cause of hyponatremia.
Hyponatremia in association with a maximally dilute urine can also result from more moderate fluid intake combined with extremely limited solute intake, a condition often referred to as beer potomania syndrome. In normal subjects, daily solute excretion is usually in the range of 800 mOsm to 1000 mOsm per day. Since the limit of urinary dilution is approximately 50 mOsm/L, then maximum urine output can be calculated to be approximately 16 L to 20 L per day (1000 mOsm/L/50 mOsm/L = 20). Individuals who drink beer or who are ingesting a fad diet have very limited protein intake so that daily solute excretion may be as low as 200 mOsm per day. In this setting, water intake >4 L/day (200 mOsm/50 mOsm/L = 4 L) will exceed renal water excretion and cause hyponatremia. Urine osmolality will be maximally dilute in such a patient.
In the absence of primary polydipsia, hyponatremia is associated with decreased renal water excretion and a urine that is inappropriately concentrated. It is important to note that in the presence of hyponatremia urine should be maximally dilute and a urine osmolality higher than this (>100 mOsm/L) is inappropriate. An inappropriately concentrated urine implies a defect in renal water excretion.
Excretion of water by the kidney is dependent on three factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be normal so that the tubular fluid will be diluted. Lastly, AVP levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is probably most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. In many conditions, decreased delivery of filtrate to the tip of the loop of Henle also contributes.
What is the Volume Status of the Patient?
In patients with hypotonic hyponatremia with an inappropriately concentrated urine, one needs to define whether effective arterial volume is decreased. Most causes of hyponatremia result from a decrease in effective arterial volume which leads to both baroreceptor‐mediated stimulation of AVP secretion and decreased distal delivery of filtrate to the tip of the loop of Henle. If effective arterial volume is low, extracellular fluid (ECF) volume can be low in the volume‐depleted patient (hypovolemic hyponatremia) or can be high in the edematous patient (hypervolemic hyponatremia). If effective arterial volume is normal, one is dealing with the euvolemic causes of hyponatremia (isovolemic hyponatremia).
The clinical determination of effective arterial volume is usually straightforward. On physical examination the best index of effective arterial volume is the presence or absence of an orthostatic change in pulse and blood pressure. Urinary electrolytes are also extremely useful in the assessment of effective arterial volume. Patients with a low effective arterial volume will tend to have a low urinary Na+ and Cl concentration and low fractional excretions of Na+ and Cl in the urine. Patients with euvolemic hyponatremia will be in balance and will excrete Na+ and Cl at rates that reflect dietary intake of Na+ and Cl. Generally urinary Na+ and Cl is >20 mEq/L and fractional excretions of these electrolytes are >1%.
Plasma composition can also be used to assess effective arterial volume. The blood urea nitrogen (BUN) is particularly sensitive to effective arterial volume. In patients with a normal serum creatinine concentration, a high BUN suggests a low effective arterial volume and a low BUN suggests a high effective arterial volume. The plasma uric acid can also be used as a sensitive index of effective arterial volume. In comparing patients with the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) and other causes of hyponatremia, patients with low effective arterial volume tend to have an elevated serum uric acid. The serum urate is low in patients with SIADH. This is due to the fact that these patients are volume expanded although it is clinically difficult to detect the degree of volume expansion.
Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
In patients who are determined to be clinically euvolemic, a concentrated urine and high AVP levels are inappropriate. The most common cause of this condition is SIADH. This syndrome is generally associated with diseases of the central nervous system, pulmonary diseases, and neoplasms. These conditions lead to secretion of AVP which is inappropriate both from the standpoint of plasma osmolality and effective arterial volume. A number of other etiologies cause a condition of hypoosmolality associated with euvolemia and can mimic the syndrome of inappropriate AVP secretion. These include isolated glucocorticoid deficiency (normal mineralocorticoid activity), hypothyroidism, pain, nausea, acute psychosis, and a variety of drugs.
Clinical Conditions Associated With Hyponatremia in the Hospital Setting
Post‐Operative Hyponatremia
The postoperative patient is particularly prone to developing hyponatremia. AVP levels are increased for several days following surgical procedures due to baroreceptor and nonbaroreceptor‐mediated mechanisms. These patients typically have subtle or overt decreases in effective arterial blood volume due to prolonged preoperative fasting combined with intraoperative and postoperative blood loss and third spacing of fluid. In addition to these factors which unload baroreceptors, postoperative pain, stress, anxiety, nausea, and administration of morphine can further stimulate the release of AVP. In some instances nonsteroidal antiinflmmatory drugs are given which have the effect of augmenting the hydroosmotic actions of AVP.6 In this setting of compromised ability to excrete a water load, administration of hypotonic fluid can precipitate acute iatrogenic hyponatremia.
Postoperative hyponatremia has been a major problem in pediatric populations due to the widespread practice of using hypotonic fluids for maintenance therapy. This approach is based on guidelines developed 50 years ago which were based on calculations linking energy expenditures to water and electrolyte losses.7 More recently, several groups have argued that isotonic rather than hypotonic fluids should be the routine maintenance fluid in such patients.8, 9 The pediatric community has been slow to embrace this approach out of the concern that excessive administration of Na+ would increase the risk of hypernatremia. A systematic meta‐analysis of studies comparing isotonic and hypotonic fluids in hospitalized children found the odds of developing hyponatremia following hypotonic solutions was 17.2 times greater than with isotonic fluids.10 The concern that isotonic maintenance fluids carry a risk of hypernatremia was not supported in the review. Some studies actually reported a decrease in serum Na+ concentration, presumably due to the desalination phenomenon in which hypertonic urine is excreted in volume expanded subjects with persistent vasopressin secretion.
Endocrine Disorders
Glucocorticoid Deficiency
Patients with glucocorticoid deficiency develop hyponatremia. It is important to separate this condition from that of mineralocorticoid deficiency and combined mineralocorticoid‐glucocorticoid deficiency. In patients with mineralocorticoid deficiency, ECF volume and effective arterial volume are low. This leads to baroreceptor stimulation of AVP secretion and to decreased distal delivery of filtrate to the diluting segments of the nephron. In isolated glucocorticoid deficiency, the patients are euvolemic. In these patients, for any given level of low plasma osmolality, vasopressin levels are inappropriately elevated. The administration of hydrocortisone restores the relationship between vasopressin and plasma osmolality to normal.
While it is possible to develop isolated glucocorticoid deficiency with adrenal disease, most adrenal diseases cause loss of mineralocorticoid and glucocorticoid function. Glucocorticoid deficiency in the absence of mineralocorticoid deficiency is usually due to pituitary disease. In fact, severe hyponatremia may be the initial clue to the presence of previously unrecognized hypopituitarism. Insufficient adrenal secretion of glucocorticoids may also be a complication following the long term use of exogenous glucocorticoids.
In addition to high AVP levels, AVP‐independent mechanisms lead to increases in urinary osmolality in glucocorticoid deficiency. The nature of this AVP‐independent effect is likely multifactorial. First, glucocorticoid deficiency is associated with a diminished cardiac output and an impaired systemic vascular response to hypotension. These changes will lead to a slight decline in glomerular filtration rate and increased volume absorption in the proximal tubule and thin descending limb. As a result, distal delivery of filtrate to the diluting segment will be abnormally low in glucocorticoid deficiency. Second, mineralocorticoid and glucocorticoid deficiency have both been shown to result in increased expression of aquaporin 2 water channels in the collecting duct.11 This later effect will further limit maximal urinary dilution and contribute to net water retention.
These AVP‐independent mechanisms are consistent with the clinical observation that patients with diabetes insipidus appear to improve clinically when they develop coexistent anterior pituitary insufficiency, and treatment of these patients with glucocorticoids appears to worsen the diabetes insipidus. Thus, in the patient with diabetes, insipidus‐free water excretion will be extremely large. When simultaneous glucocorticoid deficiency develops, free‐water excretion decreases.
Hypothyroidism
Myxedema coma is the most severe form of hypothyroidism and is commonly associated with hyponatremia.12 In this setting, blood pressure can be low because of decreased intravascular volume and cardiovascular collapse. The hypotension can be refractory to vasopressor therapy in the absence of thyroid hormone therapy. Cardiac output and stroke volume are low. A defect in renal water excretion develops as a result of baroreceptor mediated increases in AVP along with decreased delivery of filtrate to the distal nephron. In hypothyroid rats, there is upregulation of aquaporin 2 water channels. In these animals, administration of a V2 receptor antagonist reverses the increased water channel expression and corrects the impaired response to an acute water load.11
While reduced cardiac output and blood pressure associated with severe hypothyroidism can provide a stimulatory effect for AVP release through a baroreceptor mediated mechanism, milder forms of hypothyroidism can be considered in the differential diagnosis of euvolemic hyponatremia. In this setting, impaired renal excretion of water is presumably due to increased release of AVP due to the absence of a tonic inhibitory effect of thyroid hormone in the central nervous system. The degree of hyponatremia in this setting is typically mild.
Heart Failure
Hyponatremia is a common complication of left‐sided heart failure and several studies have shown that it is an independent predictor of mortality.13, 14 A similar association between reduced survival and hyponatremia is present in advanced right‐sided heart failure in patients with pulmonary arterial hypertension.15 Patients with heart failure who are hyponatremic have higher circulating levels of neurohormones (catecholamines, renin, angiotensin II, aldosterone, and AVP) than normonatremic subjects and are more likely to have prerenal azotemia. In addition to being a marker for the extent of neurohumoral activation, hyponatremia may play a more direct role in adverse outcomes through maladaptive volume regulatory responses of cardiac myocytes and by direct effects of AVP on cardiac and coronary V1a receptors.
Heart failure is associated with arterial underfilling leading to arterial baroreceptor‐mediated activation of the neurohumoral axis. This underfilling is due to decreased cardiac output in low‐output heart failure and decreased systemic vascular resistance in high‐output heart failure. Activation of the sympathetic nervous system along with the renin‐angiotensin‐aldosterone system leads to renal salt retention while the increase in AVP is associated with water retention and hyponatremia.
Cirrhosis
Hyponatremia is a common electrolyte abnormality in patients with cirrhosis and occurs with a frequency that tends to parallel the severity of liver disease.16 Patients with a serum Na+ 130 mEq/L are more likely to have refractory ascites and require therapeutic paracentesis. Hepatic encephalopathy, hepatorenal syndrome, and spontaneous bacterial peritonitis are also more common in patients with a serum Na+ 130 mmol/L than in patients with a normal serum Na+ concentration. Hyponatremia increases morbidity and mortality from hepatic transplantation and is associated with osmotic demyelination in the postoperative period because of the large increase in Na+ concentration associated with the procedure.17
Hepatic cirrhosis is characterized by a decreased effective arterial blood volume and activation of neurohumoral effectors. Reduced effective circulating volume due to generalized and specifically to arterial splanchnic vasodilation leads to baroreceptor‐mediated nonosmotic stimulation of AVP release and an impaired ability to excrete electrolyte‐free water.18 Reduced Na+ delivery to the distal tubule because of a low glomerular filtration rate and increased proximal Na+ reabsorption adds to the susceptibility of cirrhotic patients to hyponatremia.
Hyponatremic‐Hypertensive Syndrome
The development of hyponatremia in patients with severe hypertension associated with renal artery stenosis has been called the hyponatremic‐hypertensive syndrome.19, 20 Patients with this syndrome present with a variety of signs and symptoms that include headache, confusion, postural dizziness, polyuria, polydipsia, and salt craving. In a retrospective review of 32 patients with this syndrome, most of the subjects were thin, elderly, women smokers who had atherosclerotic renal vascular disease.19 Biochemical abnormalities included not only hyponatremia, but hypokalemia and increased plasma renin activity. The mean serum Na+ concentration was 129.7 mmol/L (range 120‐135).
The precise mechanism of this syndrome is not known. Given the available data, angiotensin‐mediated thirst coupled with nonosmotic release of AVP provoked by angiotensin II and/or hypertensive encephalopathy are likely. Na+ depletion due to pressure natriuresis, and K+ depletion due to hyperaldosteronism are also likely to play a role in the pathogenesis of hyponatremia.
Pneumonia
An association between pneumonia and hyponatremia has been known for quite some time but has been poorly defined. Of the various etiologic agents, legionella is more commonly associated with hyponatremia as compared to other types of community acquired disease. As has been true for many disorders, hyponatremia is associated with longer hospital stays and hospital mortality, most likely reflecting the severity of the pneumonia rather than morbidity from the usually mild and asymptomatic hyponatremia.
Central Nervous System Disease
Hyponatremia is a frequent complication of central nervous system disease to include bacterial meningitis and traumatic brain injury. Potential mechanisms include the development of SIADH, cerebral salt wasting (CSW), or hypotonic fluid administration in the setting of impaired renal water excretion as in patients with low effective volume. Of these various causes, hyponatremia is frequently attributed to SIADH. As previously mentioned, this syndrome is characterized by hyponatremia in the setting of an inappropriately concentrated urine, increased urine Na+ concentration, and evidence of normal or slightly increased intravascular volume.
However there are patients with intracranial disease who develop hyponatremia with similar characteristics but differ in that there is clinical evidence of a contracted ECF volume. This form of hyponatremia is due to excessive renal Na+ excretion resulting from a centrally mediated process and is termed CSW. The onset of this disorder is typically seen within the first ten days following a neurosurgical procedure or after a definable event, such as a subarachnoid hemorrhage or stroke. CSW has also been described in other intracranial disorders, such as carcinomatous or infectious meningitis and metastatic carcinoma.21 The distinction between SIADH and CSW is of considerable clinical importance given the divergent nature of the treatments. Fluid restriction is the treatment of choice in SIADH, whereas the treatment of CSW comprises vigorous Na+ and volume replacement.
Treatment of Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, disorientation, restlessness and obtundation, and seizures. The principal danger of hyponatremia or hypernatremia relates to effects on central nervous system function due to changes in brain size (Figure 2).
Hyponatremia initially leads to cell swelling driven by the higher intracellular osmolality. The net result is equilibration of intracellular and extracellular osmolality at the expense of increased brain volume. Cells in general, and brain cells in particular, then respond by decreasing the number of intracellular osmoles and as intracellular osmolality decreases, cell size returns toward normal despite the presence of hyponatremia. If the decrease in ECF osmolality is slow, there will be no measurable cell swelling. This pathophysiologic sequence correlates well with clinical observations. If hyponatremia is slow in onset, neurologic symptoms and permanent brain damage are unusual, even if the decreases in Na+ concentration and ECF osmolality are large. Conversely, if hyponatremia is rapid in onset, cerebral edema and significant CNS symptoms and signs can occur with lesser changes in serum Na+ concentration.
When treating a patient with hyponatremia, the Na+ concentration should be raised at the rate at which it fell. In a patient whose serum Na+ concentration has decreased rapidly (<48 hours), neurologic symptoms are frequently present and there is cerebral edema. In this setting there has not been sufficient time to remove osmoles from the brain and rapid return to normal ECF osmolality merely returns brain size to normal. In general, the development of hyponatremia in the outpatient setting is more commonly chronic in duration and should be corrected slowly. By contrast, hyponatremia of short duration is more likely to be encountered in hospitalized patients receiving intravenous free water. Use of ecstasy, exercise‐induced hyponatremia, or patients with primary polydipsia can also develop acute hyponatremia and if symptomatic may similarly require rapid correction. Raising the serum Na+ concentration by 4 mEq/L to 6 mEq/L over a several hour period is both safe and effective in preventing untoward effects of acute hyponatremia.22 A reasonable strategy to accomplish this goal is the regimen recommended for the treatment of athletes with hyponatremia and encephalopathy.23 These guidelines suggest an immediate bolus of 100 mL 3% NaCl (513 mEq/L). If there is no neurologic improvement two additional 100 mL 3% NaCl bolus infusions separated by 10 minute intervals can be given.
In patients with chronic hyponatremia (>48 hours duration) the serum Na+ concentration has fallen slowly. Neurologic symptoms are generally minimal, brain size is normal, and the number of intracellular osmoles is decreased. Sudden return of ECF osmolality to normal values will lead to cell shrinkage and possibly precipitate osmotic demyelination. Experts in the field suggest this complication can be prevented by adhering to the following limits of correction: 10 mmol/L in 24 hours, 18 mmol/L in 48 hours, and 20 mmol/L in 72 hours.22 In order to maximize patient safety, the goals of therapy should be more modest: 6 to 8 mmol/L in 24 hours, 12 to 14 mmol/L in 48 hours, and 14 to 16 mmol/L in 72 hours.
A formula designed to predict the increase in serum Na+ concentration to be expected from the infusion of 1 L of a given infusion is given below:
| Treament | Mechanism | Advantages | Limitations |
|---|---|---|---|
| |||
| Fluid restriction (0.5‐1.0 L/day) | water intake | Effective, inexpensive | Poor compliance |
| Demeclocyline (600‐1200 mg/day) | Inhibits action of AVP | Widely available | Slow onset, nephrotoxic, expensive |
| Urea (30 gm/day) | Osmotic diuresis | Possiblerisk of osmotic demyelination | Poor palatability, avoid in chronic kidney and liver disease |
| Lithium (up to 900 mg/daily) | Inhibits action of AVP | Widely available | Slow onset, toxicity |
When treating patients with SIADH with intravenous salt solutions, one can worsen the degree of hyponatremia if the osmolality of the administered fluid is less that the urine osmolality. One liter of isotonic saline contains 150 mEq each of Na+ and Cl and has an osmolality of 300 mOsm. In a patient with SIADH who has a urine osmolality of 600 mOsm, the administered NaCl will all be excreted in 500 mL of water (300 mOsm in 500 mL = 600 mOsm/L). The remaining 500 mL of administered fluid will be retained in the body and cause further dilution of the serum Na+ concentration. By contrast, 1 L of 3% saline contains 513 mEq each of Na+ and Cl and has an osmolality of 1026 mOsm. All of the NaCl in 1 L of this solution will be excreted in 1.7 L of water and therefore will cause the serum Na+ concentration to increase as a result of the loss of 700 mL of water.
Loop diuretics can also be employed as part of the strategy to treat chronic hyponatremia. Loop diuretics directly interfere in the countercurrent concentrating mechanism by inhibiting NaCl reabsorption in the loop of Henle. The coadministration of these drugs will enhance the effect of intravenous salt solutions by lowering urine osmolality thereby increasing the likelihood that the osmolality of the administered fluid is greater than the urine osmolality. Other measures used to treat chronic hyponatremia are given in Table 1.
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of water channels (aquaporin 2) into the apical membrane. The net effect is increased water excretion with little to no change in urinary Na+ excretion. AVP antagonists have been shown to be more effective than placebo in increasing the serum Na+ concentration in patients with modest asymptomatic hyponatremia.26, 27 These agents can be considered in patients with hyponatremia associated with heart failure or cirrhosis and in patients with irreversible SIADH but should not be used in patients with hypovolemic hyponatremia. The role of these drugs is discussed further in an accompanying article in this supplement, titled New horizons in the pharmacologic approach to hyponatremia: The V2 Receptor Antagonists.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chem Acta.2003;337:169–172.
- ,,,.Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin.Ann Intern Med.1985;102(2):164–168.
- ,,, et al.Characteristics and mortality of severe hyponatraemia‐a hospital‐based study.Clin Endocrinol (Oxf).2006;65(2):246–249.
- ,,.Mortality after hospitalization with mild moderate and severe hyponatremia.Am J Med.2009;122:8857–8865.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- .Renal complications associated with use of nonsteroidal anti‐inflammatory agents.J Investig Med.1995;43(6):516–533.
- ,.Recent developments in the perioperative fluid management for the paediatric patient.Curr Opin Anaesthesiol.2006;19:268–277.
- ,.Prevention of hospital‐acquired hyponatremia: a case for using isotonic saline.Pediatrics.2003;111:227–230.
- ,,,,.Acute hyponatremia related to intravenous fluid administration in hospitalized children: an observational study.Pediatrics.2004;113(5):1279–1284.
- ,,,.Hypotonic versus isotonic saline in hospitalized children: a systematic review.Arch Dis Child.2006;91(10):828–835.
- .Vasopressin and Aquaporin 2 in clinical disorders of water homeostasis.Semin Nephrol.2008;28:289–296.
- .Myxedema coma.Edocrinol Metab Clin N Am.2006;35:687–698.
- ,,, et al.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28(8):920–921.
- ,.Pulmonary hypertension, right ventricular failure, and kidney: different from left ventricular failure.Clin J Am Soc Nephrol.2008;3:1232–1237.
- ,,, et al.Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension.Am J Respir Crit Care Med.2008;177(12):1364–1369.
- ,,,;CAPPS investigators. Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,,,,.Possible causes of central pontine myelinolysis after liver transplantation.World J Gastroenterol.2004;10(17):2540–2543.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.The hyponatremic hypertensive syndrome in renal artery stenosis: An infrequent cause of hyponatremia.J Postgrad Med.2007;53:41–43.
- ,,,.Hyponatremic‐hypertensive syndrome with renal ischemia: an underrecognized disorder.Hypertension.1993;33:1020–1024.
- .Hyponatremia in patients with central nervous system disease: SIADH or CSW.Trends Endocrinol Metab.2003;14:182–187.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- ,,, et al.Statement of the second international exercise‐associated hyponatremia consensus development conference, New Zealand.Clin J Sport Med.2008;18:111–121.
- ,,,,,.Hypertonic saline for hyponatremia: risk of inadvertent overcorrection.Clin J Am Soc Nephrol.2007;2:1110–1117.
- ,,, et al.DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia.Clin J Am Soc Nephrol.2008;3:331–336.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol2007;27:447–457.
- ,,, et al.,Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
Hyponatremia is one of the most common electrolyte abnormalities encountered in clinical practice. The frequency of the disorder varies according to definition and clinical setting but has been reported to be present in 28% of patients upon hospital admission and in 7% of patients attending an ambulatory community clinic.1 Increasing age, medications, various disease states, and administration of hypotonic fluids are among the known risk factors for the disorder.
The mortality rate in hyponatremic patients is approximately 3 times that of normonatremic hospitalized patients.25 Outcomes are particularly poor in those patients whose serum sodium (Na+) falls during a hospitalization. In 1 prospective study the mortality rate in patients with a normal serum Na+ concentration was 0.2% in comparison to a mortality rate of 11.2% and 25% in patients with a serum Na+ concentration <130 mEq/L and <120 mEq/L, respectively.2 In a recent retrospective cohort study of 10,899 hospitalized patients, the incidence of hyponatremia (<135 mmol/L) at admission was 5.5%.5 As compared to those with normonatremia, these patients were more likely to require intensive care and mechanical ventilation within 48 hours of hospitalization. In addition, hospital mortality, mean length of stay, and costs were significantly greater among patients with hyponatremia than those without.
The association with hyponatremia and adverse outcomes could be the direct result of hyponatremia, the comormidities that lead to the electrolyte derangement, or both. Whatever the mechanism, hyponatremia should not be viewed as an innocuous condition. Rather, clinicians should view this disorder with urgency and institute measures to prevent any further decline in the serum Na+ concentration and initiate appropriate therapy for its correction. This review will first briefly summarize the pathogenesis of hyponatremia and then discuss various disease states encountered in the hospital setting in which hyponatremia is frequently present.
Pathogenesis of Hyponatremia
Hyponatremia is generally associated with a hypoosmolar state and is a marker for a disturbance in water balance. Stated differently, all hyponatremia is dilutional. The approach to the patient with hyponatremia is outlined in Figure 1.
Is the Hyponatremia Representative of a Hypoosmolar State?
There are 3 causes of hyponatremia in which it is not associated with a hypoosmolar state. The first of these is pseudohyponatremia which involves an abnormal measurement of the serum Na+. This occurs in patients with hyperglobulinemia or hypertriglyceridemia in whom plasma water relative to plasma solids is decreased in blood, leading to less Na+ in a given volume of blood. In general, this problem is becoming less prevalent as many laboratories are using Na+ electrodes without diluting the blood such that the Na+ measurement becomes independent of plasma water and nonaqueous contents.
A second cause of hyponatremia in the absence of a hypoosmolar state involves true hyponatremia but with elevations in the concentration of another osmole. Clinical examples include hyperglycemia as seen in uncontrolled diabetes or rarely hypertonic infusion of mannitol used in the treatment of cerebral edema. The accumulation of these effective osmoles creates an osmotic force causing water to move from the intracellular to the extracellular space thus diluting the serum Na+. For every 100 mg/dL rise in glucose or mannitol, the serum Na+ will quickly fall by 1.6 mEq/L. This increase in tonicity also stimulates thirst and arginine vasopressin (AVP) secretion, both of which contribute to further water retention. As a result the plasma osmolality and serum Na+ concentration will continue to fall. Once the plasma osmolality normalizes the serum Na+ will have decreased by 2.8 mEq/L for every 100 mg/dL rise in glucose.
The third cause of hyponatremia in the absence of a hypoosmolar state is the addition of an isosmotic (or near isosmotic) non‐Na+ containing fluid to the extracellular space. This situation typically occurs during a transurethral resection of the prostate or during laprascopic surgery when large amounts of a nonconducting flushing solution containing glycine or sorbitol are absorbed systemically.
Is the Kidney's Ability to Dilute the Urine Intact?
The presence of hypotonic hyponatremia implies that water intake exceeds the ability of the kidney to excrete water. In unusual circumstances, this can occur when the kidneys ability to excrete free water is intact. However, because a normal kidney can excrete 18 L of water per day, the presence of hyponatremia with normal renal water excretion implies the patient is drinking >20 L water/day. This condition is referred to as primary polydipsia. These patients should have a urine osmolality <100 mOsm/L. While primary polydipsia is a common condition which leads to polyuria and polydipsia, it is uncommon as a sole cause of hyponatremia.
Hyponatremia in association with a maximally dilute urine can also result from more moderate fluid intake combined with extremely limited solute intake, a condition often referred to as beer potomania syndrome. In normal subjects, daily solute excretion is usually in the range of 800 mOsm to 1000 mOsm per day. Since the limit of urinary dilution is approximately 50 mOsm/L, then maximum urine output can be calculated to be approximately 16 L to 20 L per day (1000 mOsm/L/50 mOsm/L = 20). Individuals who drink beer or who are ingesting a fad diet have very limited protein intake so that daily solute excretion may be as low as 200 mOsm per day. In this setting, water intake >4 L/day (200 mOsm/50 mOsm/L = 4 L) will exceed renal water excretion and cause hyponatremia. Urine osmolality will be maximally dilute in such a patient.
In the absence of primary polydipsia, hyponatremia is associated with decreased renal water excretion and a urine that is inappropriately concentrated. It is important to note that in the presence of hyponatremia urine should be maximally dilute and a urine osmolality higher than this (>100 mOsm/L) is inappropriate. An inappropriately concentrated urine implies a defect in renal water excretion.
Excretion of water by the kidney is dependent on three factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be normal so that the tubular fluid will be diluted. Lastly, AVP levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is probably most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. In many conditions, decreased delivery of filtrate to the tip of the loop of Henle also contributes.
What is the Volume Status of the Patient?
In patients with hypotonic hyponatremia with an inappropriately concentrated urine, one needs to define whether effective arterial volume is decreased. Most causes of hyponatremia result from a decrease in effective arterial volume which leads to both baroreceptor‐mediated stimulation of AVP secretion and decreased distal delivery of filtrate to the tip of the loop of Henle. If effective arterial volume is low, extracellular fluid (ECF) volume can be low in the volume‐depleted patient (hypovolemic hyponatremia) or can be high in the edematous patient (hypervolemic hyponatremia). If effective arterial volume is normal, one is dealing with the euvolemic causes of hyponatremia (isovolemic hyponatremia).
The clinical determination of effective arterial volume is usually straightforward. On physical examination the best index of effective arterial volume is the presence or absence of an orthostatic change in pulse and blood pressure. Urinary electrolytes are also extremely useful in the assessment of effective arterial volume. Patients with a low effective arterial volume will tend to have a low urinary Na+ and Cl concentration and low fractional excretions of Na+ and Cl in the urine. Patients with euvolemic hyponatremia will be in balance and will excrete Na+ and Cl at rates that reflect dietary intake of Na+ and Cl. Generally urinary Na+ and Cl is >20 mEq/L and fractional excretions of these electrolytes are >1%.
Plasma composition can also be used to assess effective arterial volume. The blood urea nitrogen (BUN) is particularly sensitive to effective arterial volume. In patients with a normal serum creatinine concentration, a high BUN suggests a low effective arterial volume and a low BUN suggests a high effective arterial volume. The plasma uric acid can also be used as a sensitive index of effective arterial volume. In comparing patients with the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) and other causes of hyponatremia, patients with low effective arterial volume tend to have an elevated serum uric acid. The serum urate is low in patients with SIADH. This is due to the fact that these patients are volume expanded although it is clinically difficult to detect the degree of volume expansion.
Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
In patients who are determined to be clinically euvolemic, a concentrated urine and high AVP levels are inappropriate. The most common cause of this condition is SIADH. This syndrome is generally associated with diseases of the central nervous system, pulmonary diseases, and neoplasms. These conditions lead to secretion of AVP which is inappropriate both from the standpoint of plasma osmolality and effective arterial volume. A number of other etiologies cause a condition of hypoosmolality associated with euvolemia and can mimic the syndrome of inappropriate AVP secretion. These include isolated glucocorticoid deficiency (normal mineralocorticoid activity), hypothyroidism, pain, nausea, acute psychosis, and a variety of drugs.
Clinical Conditions Associated With Hyponatremia in the Hospital Setting
Post‐Operative Hyponatremia
The postoperative patient is particularly prone to developing hyponatremia. AVP levels are increased for several days following surgical procedures due to baroreceptor and nonbaroreceptor‐mediated mechanisms. These patients typically have subtle or overt decreases in effective arterial blood volume due to prolonged preoperative fasting combined with intraoperative and postoperative blood loss and third spacing of fluid. In addition to these factors which unload baroreceptors, postoperative pain, stress, anxiety, nausea, and administration of morphine can further stimulate the release of AVP. In some instances nonsteroidal antiinflmmatory drugs are given which have the effect of augmenting the hydroosmotic actions of AVP.6 In this setting of compromised ability to excrete a water load, administration of hypotonic fluid can precipitate acute iatrogenic hyponatremia.
Postoperative hyponatremia has been a major problem in pediatric populations due to the widespread practice of using hypotonic fluids for maintenance therapy. This approach is based on guidelines developed 50 years ago which were based on calculations linking energy expenditures to water and electrolyte losses.7 More recently, several groups have argued that isotonic rather than hypotonic fluids should be the routine maintenance fluid in such patients.8, 9 The pediatric community has been slow to embrace this approach out of the concern that excessive administration of Na+ would increase the risk of hypernatremia. A systematic meta‐analysis of studies comparing isotonic and hypotonic fluids in hospitalized children found the odds of developing hyponatremia following hypotonic solutions was 17.2 times greater than with isotonic fluids.10 The concern that isotonic maintenance fluids carry a risk of hypernatremia was not supported in the review. Some studies actually reported a decrease in serum Na+ concentration, presumably due to the desalination phenomenon in which hypertonic urine is excreted in volume expanded subjects with persistent vasopressin secretion.
Endocrine Disorders
Glucocorticoid Deficiency
Patients with glucocorticoid deficiency develop hyponatremia. It is important to separate this condition from that of mineralocorticoid deficiency and combined mineralocorticoid‐glucocorticoid deficiency. In patients with mineralocorticoid deficiency, ECF volume and effective arterial volume are low. This leads to baroreceptor stimulation of AVP secretion and to decreased distal delivery of filtrate to the diluting segments of the nephron. In isolated glucocorticoid deficiency, the patients are euvolemic. In these patients, for any given level of low plasma osmolality, vasopressin levels are inappropriately elevated. The administration of hydrocortisone restores the relationship between vasopressin and plasma osmolality to normal.
While it is possible to develop isolated glucocorticoid deficiency with adrenal disease, most adrenal diseases cause loss of mineralocorticoid and glucocorticoid function. Glucocorticoid deficiency in the absence of mineralocorticoid deficiency is usually due to pituitary disease. In fact, severe hyponatremia may be the initial clue to the presence of previously unrecognized hypopituitarism. Insufficient adrenal secretion of glucocorticoids may also be a complication following the long term use of exogenous glucocorticoids.
In addition to high AVP levels, AVP‐independent mechanisms lead to increases in urinary osmolality in glucocorticoid deficiency. The nature of this AVP‐independent effect is likely multifactorial. First, glucocorticoid deficiency is associated with a diminished cardiac output and an impaired systemic vascular response to hypotension. These changes will lead to a slight decline in glomerular filtration rate and increased volume absorption in the proximal tubule and thin descending limb. As a result, distal delivery of filtrate to the diluting segment will be abnormally low in glucocorticoid deficiency. Second, mineralocorticoid and glucocorticoid deficiency have both been shown to result in increased expression of aquaporin 2 water channels in the collecting duct.11 This later effect will further limit maximal urinary dilution and contribute to net water retention.
These AVP‐independent mechanisms are consistent with the clinical observation that patients with diabetes insipidus appear to improve clinically when they develop coexistent anterior pituitary insufficiency, and treatment of these patients with glucocorticoids appears to worsen the diabetes insipidus. Thus, in the patient with diabetes, insipidus‐free water excretion will be extremely large. When simultaneous glucocorticoid deficiency develops, free‐water excretion decreases.
Hypothyroidism
Myxedema coma is the most severe form of hypothyroidism and is commonly associated with hyponatremia.12 In this setting, blood pressure can be low because of decreased intravascular volume and cardiovascular collapse. The hypotension can be refractory to vasopressor therapy in the absence of thyroid hormone therapy. Cardiac output and stroke volume are low. A defect in renal water excretion develops as a result of baroreceptor mediated increases in AVP along with decreased delivery of filtrate to the distal nephron. In hypothyroid rats, there is upregulation of aquaporin 2 water channels. In these animals, administration of a V2 receptor antagonist reverses the increased water channel expression and corrects the impaired response to an acute water load.11
While reduced cardiac output and blood pressure associated with severe hypothyroidism can provide a stimulatory effect for AVP release through a baroreceptor mediated mechanism, milder forms of hypothyroidism can be considered in the differential diagnosis of euvolemic hyponatremia. In this setting, impaired renal excretion of water is presumably due to increased release of AVP due to the absence of a tonic inhibitory effect of thyroid hormone in the central nervous system. The degree of hyponatremia in this setting is typically mild.
Heart Failure
Hyponatremia is a common complication of left‐sided heart failure and several studies have shown that it is an independent predictor of mortality.13, 14 A similar association between reduced survival and hyponatremia is present in advanced right‐sided heart failure in patients with pulmonary arterial hypertension.15 Patients with heart failure who are hyponatremic have higher circulating levels of neurohormones (catecholamines, renin, angiotensin II, aldosterone, and AVP) than normonatremic subjects and are more likely to have prerenal azotemia. In addition to being a marker for the extent of neurohumoral activation, hyponatremia may play a more direct role in adverse outcomes through maladaptive volume regulatory responses of cardiac myocytes and by direct effects of AVP on cardiac and coronary V1a receptors.
Heart failure is associated with arterial underfilling leading to arterial baroreceptor‐mediated activation of the neurohumoral axis. This underfilling is due to decreased cardiac output in low‐output heart failure and decreased systemic vascular resistance in high‐output heart failure. Activation of the sympathetic nervous system along with the renin‐angiotensin‐aldosterone system leads to renal salt retention while the increase in AVP is associated with water retention and hyponatremia.
Cirrhosis
Hyponatremia is a common electrolyte abnormality in patients with cirrhosis and occurs with a frequency that tends to parallel the severity of liver disease.16 Patients with a serum Na+ 130 mEq/L are more likely to have refractory ascites and require therapeutic paracentesis. Hepatic encephalopathy, hepatorenal syndrome, and spontaneous bacterial peritonitis are also more common in patients with a serum Na+ 130 mmol/L than in patients with a normal serum Na+ concentration. Hyponatremia increases morbidity and mortality from hepatic transplantation and is associated with osmotic demyelination in the postoperative period because of the large increase in Na+ concentration associated with the procedure.17
Hepatic cirrhosis is characterized by a decreased effective arterial blood volume and activation of neurohumoral effectors. Reduced effective circulating volume due to generalized and specifically to arterial splanchnic vasodilation leads to baroreceptor‐mediated nonosmotic stimulation of AVP release and an impaired ability to excrete electrolyte‐free water.18 Reduced Na+ delivery to the distal tubule because of a low glomerular filtration rate and increased proximal Na+ reabsorption adds to the susceptibility of cirrhotic patients to hyponatremia.
Hyponatremic‐Hypertensive Syndrome
The development of hyponatremia in patients with severe hypertension associated with renal artery stenosis has been called the hyponatremic‐hypertensive syndrome.19, 20 Patients with this syndrome present with a variety of signs and symptoms that include headache, confusion, postural dizziness, polyuria, polydipsia, and salt craving. In a retrospective review of 32 patients with this syndrome, most of the subjects were thin, elderly, women smokers who had atherosclerotic renal vascular disease.19 Biochemical abnormalities included not only hyponatremia, but hypokalemia and increased plasma renin activity. The mean serum Na+ concentration was 129.7 mmol/L (range 120‐135).
The precise mechanism of this syndrome is not known. Given the available data, angiotensin‐mediated thirst coupled with nonosmotic release of AVP provoked by angiotensin II and/or hypertensive encephalopathy are likely. Na+ depletion due to pressure natriuresis, and K+ depletion due to hyperaldosteronism are also likely to play a role in the pathogenesis of hyponatremia.
Pneumonia
An association between pneumonia and hyponatremia has been known for quite some time but has been poorly defined. Of the various etiologic agents, legionella is more commonly associated with hyponatremia as compared to other types of community acquired disease. As has been true for many disorders, hyponatremia is associated with longer hospital stays and hospital mortality, most likely reflecting the severity of the pneumonia rather than morbidity from the usually mild and asymptomatic hyponatremia.
Central Nervous System Disease
Hyponatremia is a frequent complication of central nervous system disease to include bacterial meningitis and traumatic brain injury. Potential mechanisms include the development of SIADH, cerebral salt wasting (CSW), or hypotonic fluid administration in the setting of impaired renal water excretion as in patients with low effective volume. Of these various causes, hyponatremia is frequently attributed to SIADH. As previously mentioned, this syndrome is characterized by hyponatremia in the setting of an inappropriately concentrated urine, increased urine Na+ concentration, and evidence of normal or slightly increased intravascular volume.
However there are patients with intracranial disease who develop hyponatremia with similar characteristics but differ in that there is clinical evidence of a contracted ECF volume. This form of hyponatremia is due to excessive renal Na+ excretion resulting from a centrally mediated process and is termed CSW. The onset of this disorder is typically seen within the first ten days following a neurosurgical procedure or after a definable event, such as a subarachnoid hemorrhage or stroke. CSW has also been described in other intracranial disorders, such as carcinomatous or infectious meningitis and metastatic carcinoma.21 The distinction between SIADH and CSW is of considerable clinical importance given the divergent nature of the treatments. Fluid restriction is the treatment of choice in SIADH, whereas the treatment of CSW comprises vigorous Na+ and volume replacement.
Treatment of Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, disorientation, restlessness and obtundation, and seizures. The principal danger of hyponatremia or hypernatremia relates to effects on central nervous system function due to changes in brain size (Figure 2).
Hyponatremia initially leads to cell swelling driven by the higher intracellular osmolality. The net result is equilibration of intracellular and extracellular osmolality at the expense of increased brain volume. Cells in general, and brain cells in particular, then respond by decreasing the number of intracellular osmoles and as intracellular osmolality decreases, cell size returns toward normal despite the presence of hyponatremia. If the decrease in ECF osmolality is slow, there will be no measurable cell swelling. This pathophysiologic sequence correlates well with clinical observations. If hyponatremia is slow in onset, neurologic symptoms and permanent brain damage are unusual, even if the decreases in Na+ concentration and ECF osmolality are large. Conversely, if hyponatremia is rapid in onset, cerebral edema and significant CNS symptoms and signs can occur with lesser changes in serum Na+ concentration.
When treating a patient with hyponatremia, the Na+ concentration should be raised at the rate at which it fell. In a patient whose serum Na+ concentration has decreased rapidly (<48 hours), neurologic symptoms are frequently present and there is cerebral edema. In this setting there has not been sufficient time to remove osmoles from the brain and rapid return to normal ECF osmolality merely returns brain size to normal. In general, the development of hyponatremia in the outpatient setting is more commonly chronic in duration and should be corrected slowly. By contrast, hyponatremia of short duration is more likely to be encountered in hospitalized patients receiving intravenous free water. Use of ecstasy, exercise‐induced hyponatremia, or patients with primary polydipsia can also develop acute hyponatremia and if symptomatic may similarly require rapid correction. Raising the serum Na+ concentration by 4 mEq/L to 6 mEq/L over a several hour period is both safe and effective in preventing untoward effects of acute hyponatremia.22 A reasonable strategy to accomplish this goal is the regimen recommended for the treatment of athletes with hyponatremia and encephalopathy.23 These guidelines suggest an immediate bolus of 100 mL 3% NaCl (513 mEq/L). If there is no neurologic improvement two additional 100 mL 3% NaCl bolus infusions separated by 10 minute intervals can be given.
In patients with chronic hyponatremia (>48 hours duration) the serum Na+ concentration has fallen slowly. Neurologic symptoms are generally minimal, brain size is normal, and the number of intracellular osmoles is decreased. Sudden return of ECF osmolality to normal values will lead to cell shrinkage and possibly precipitate osmotic demyelination. Experts in the field suggest this complication can be prevented by adhering to the following limits of correction: 10 mmol/L in 24 hours, 18 mmol/L in 48 hours, and 20 mmol/L in 72 hours.22 In order to maximize patient safety, the goals of therapy should be more modest: 6 to 8 mmol/L in 24 hours, 12 to 14 mmol/L in 48 hours, and 14 to 16 mmol/L in 72 hours.
A formula designed to predict the increase in serum Na+ concentration to be expected from the infusion of 1 L of a given infusion is given below:
| Treament | Mechanism | Advantages | Limitations |
|---|---|---|---|
| |||
| Fluid restriction (0.5‐1.0 L/day) | water intake | Effective, inexpensive | Poor compliance |
| Demeclocyline (600‐1200 mg/day) | Inhibits action of AVP | Widely available | Slow onset, nephrotoxic, expensive |
| Urea (30 gm/day) | Osmotic diuresis | Possiblerisk of osmotic demyelination | Poor palatability, avoid in chronic kidney and liver disease |
| Lithium (up to 900 mg/daily) | Inhibits action of AVP | Widely available | Slow onset, toxicity |
When treating patients with SIADH with intravenous salt solutions, one can worsen the degree of hyponatremia if the osmolality of the administered fluid is less that the urine osmolality. One liter of isotonic saline contains 150 mEq each of Na+ and Cl and has an osmolality of 300 mOsm. In a patient with SIADH who has a urine osmolality of 600 mOsm, the administered NaCl will all be excreted in 500 mL of water (300 mOsm in 500 mL = 600 mOsm/L). The remaining 500 mL of administered fluid will be retained in the body and cause further dilution of the serum Na+ concentration. By contrast, 1 L of 3% saline contains 513 mEq each of Na+ and Cl and has an osmolality of 1026 mOsm. All of the NaCl in 1 L of this solution will be excreted in 1.7 L of water and therefore will cause the serum Na+ concentration to increase as a result of the loss of 700 mL of water.
Loop diuretics can also be employed as part of the strategy to treat chronic hyponatremia. Loop diuretics directly interfere in the countercurrent concentrating mechanism by inhibiting NaCl reabsorption in the loop of Henle. The coadministration of these drugs will enhance the effect of intravenous salt solutions by lowering urine osmolality thereby increasing the likelihood that the osmolality of the administered fluid is greater than the urine osmolality. Other measures used to treat chronic hyponatremia are given in Table 1.
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of water channels (aquaporin 2) into the apical membrane. The net effect is increased water excretion with little to no change in urinary Na+ excretion. AVP antagonists have been shown to be more effective than placebo in increasing the serum Na+ concentration in patients with modest asymptomatic hyponatremia.26, 27 These agents can be considered in patients with hyponatremia associated with heart failure or cirrhosis and in patients with irreversible SIADH but should not be used in patients with hypovolemic hyponatremia. The role of these drugs is discussed further in an accompanying article in this supplement, titled New horizons in the pharmacologic approach to hyponatremia: The V2 Receptor Antagonists.
Hyponatremia is one of the most common electrolyte abnormalities encountered in clinical practice. The frequency of the disorder varies according to definition and clinical setting but has been reported to be present in 28% of patients upon hospital admission and in 7% of patients attending an ambulatory community clinic.1 Increasing age, medications, various disease states, and administration of hypotonic fluids are among the known risk factors for the disorder.
The mortality rate in hyponatremic patients is approximately 3 times that of normonatremic hospitalized patients.25 Outcomes are particularly poor in those patients whose serum sodium (Na+) falls during a hospitalization. In 1 prospective study the mortality rate in patients with a normal serum Na+ concentration was 0.2% in comparison to a mortality rate of 11.2% and 25% in patients with a serum Na+ concentration <130 mEq/L and <120 mEq/L, respectively.2 In a recent retrospective cohort study of 10,899 hospitalized patients, the incidence of hyponatremia (<135 mmol/L) at admission was 5.5%.5 As compared to those with normonatremia, these patients were more likely to require intensive care and mechanical ventilation within 48 hours of hospitalization. In addition, hospital mortality, mean length of stay, and costs were significantly greater among patients with hyponatremia than those without.
The association with hyponatremia and adverse outcomes could be the direct result of hyponatremia, the comormidities that lead to the electrolyte derangement, or both. Whatever the mechanism, hyponatremia should not be viewed as an innocuous condition. Rather, clinicians should view this disorder with urgency and institute measures to prevent any further decline in the serum Na+ concentration and initiate appropriate therapy for its correction. This review will first briefly summarize the pathogenesis of hyponatremia and then discuss various disease states encountered in the hospital setting in which hyponatremia is frequently present.
Pathogenesis of Hyponatremia
Hyponatremia is generally associated with a hypoosmolar state and is a marker for a disturbance in water balance. Stated differently, all hyponatremia is dilutional. The approach to the patient with hyponatremia is outlined in Figure 1.
Is the Hyponatremia Representative of a Hypoosmolar State?
There are 3 causes of hyponatremia in which it is not associated with a hypoosmolar state. The first of these is pseudohyponatremia which involves an abnormal measurement of the serum Na+. This occurs in patients with hyperglobulinemia or hypertriglyceridemia in whom plasma water relative to plasma solids is decreased in blood, leading to less Na+ in a given volume of blood. In general, this problem is becoming less prevalent as many laboratories are using Na+ electrodes without diluting the blood such that the Na+ measurement becomes independent of plasma water and nonaqueous contents.
A second cause of hyponatremia in the absence of a hypoosmolar state involves true hyponatremia but with elevations in the concentration of another osmole. Clinical examples include hyperglycemia as seen in uncontrolled diabetes or rarely hypertonic infusion of mannitol used in the treatment of cerebral edema. The accumulation of these effective osmoles creates an osmotic force causing water to move from the intracellular to the extracellular space thus diluting the serum Na+. For every 100 mg/dL rise in glucose or mannitol, the serum Na+ will quickly fall by 1.6 mEq/L. This increase in tonicity also stimulates thirst and arginine vasopressin (AVP) secretion, both of which contribute to further water retention. As a result the plasma osmolality and serum Na+ concentration will continue to fall. Once the plasma osmolality normalizes the serum Na+ will have decreased by 2.8 mEq/L for every 100 mg/dL rise in glucose.
The third cause of hyponatremia in the absence of a hypoosmolar state is the addition of an isosmotic (or near isosmotic) non‐Na+ containing fluid to the extracellular space. This situation typically occurs during a transurethral resection of the prostate or during laprascopic surgery when large amounts of a nonconducting flushing solution containing glycine or sorbitol are absorbed systemically.
Is the Kidney's Ability to Dilute the Urine Intact?
The presence of hypotonic hyponatremia implies that water intake exceeds the ability of the kidney to excrete water. In unusual circumstances, this can occur when the kidneys ability to excrete free water is intact. However, because a normal kidney can excrete 18 L of water per day, the presence of hyponatremia with normal renal water excretion implies the patient is drinking >20 L water/day. This condition is referred to as primary polydipsia. These patients should have a urine osmolality <100 mOsm/L. While primary polydipsia is a common condition which leads to polyuria and polydipsia, it is uncommon as a sole cause of hyponatremia.
Hyponatremia in association with a maximally dilute urine can also result from more moderate fluid intake combined with extremely limited solute intake, a condition often referred to as beer potomania syndrome. In normal subjects, daily solute excretion is usually in the range of 800 mOsm to 1000 mOsm per day. Since the limit of urinary dilution is approximately 50 mOsm/L, then maximum urine output can be calculated to be approximately 16 L to 20 L per day (1000 mOsm/L/50 mOsm/L = 20). Individuals who drink beer or who are ingesting a fad diet have very limited protein intake so that daily solute excretion may be as low as 200 mOsm per day. In this setting, water intake >4 L/day (200 mOsm/50 mOsm/L = 4 L) will exceed renal water excretion and cause hyponatremia. Urine osmolality will be maximally dilute in such a patient.
In the absence of primary polydipsia, hyponatremia is associated with decreased renal water excretion and a urine that is inappropriately concentrated. It is important to note that in the presence of hyponatremia urine should be maximally dilute and a urine osmolality higher than this (>100 mOsm/L) is inappropriate. An inappropriately concentrated urine implies a defect in renal water excretion.
Excretion of water by the kidney is dependent on three factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be normal so that the tubular fluid will be diluted. Lastly, AVP levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is probably most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. In many conditions, decreased delivery of filtrate to the tip of the loop of Henle also contributes.
What is the Volume Status of the Patient?
In patients with hypotonic hyponatremia with an inappropriately concentrated urine, one needs to define whether effective arterial volume is decreased. Most causes of hyponatremia result from a decrease in effective arterial volume which leads to both baroreceptor‐mediated stimulation of AVP secretion and decreased distal delivery of filtrate to the tip of the loop of Henle. If effective arterial volume is low, extracellular fluid (ECF) volume can be low in the volume‐depleted patient (hypovolemic hyponatremia) or can be high in the edematous patient (hypervolemic hyponatremia). If effective arterial volume is normal, one is dealing with the euvolemic causes of hyponatremia (isovolemic hyponatremia).
The clinical determination of effective arterial volume is usually straightforward. On physical examination the best index of effective arterial volume is the presence or absence of an orthostatic change in pulse and blood pressure. Urinary electrolytes are also extremely useful in the assessment of effective arterial volume. Patients with a low effective arterial volume will tend to have a low urinary Na+ and Cl concentration and low fractional excretions of Na+ and Cl in the urine. Patients with euvolemic hyponatremia will be in balance and will excrete Na+ and Cl at rates that reflect dietary intake of Na+ and Cl. Generally urinary Na+ and Cl is >20 mEq/L and fractional excretions of these electrolytes are >1%.
Plasma composition can also be used to assess effective arterial volume. The blood urea nitrogen (BUN) is particularly sensitive to effective arterial volume. In patients with a normal serum creatinine concentration, a high BUN suggests a low effective arterial volume and a low BUN suggests a high effective arterial volume. The plasma uric acid can also be used as a sensitive index of effective arterial volume. In comparing patients with the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) and other causes of hyponatremia, patients with low effective arterial volume tend to have an elevated serum uric acid. The serum urate is low in patients with SIADH. This is due to the fact that these patients are volume expanded although it is clinically difficult to detect the degree of volume expansion.
Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
In patients who are determined to be clinically euvolemic, a concentrated urine and high AVP levels are inappropriate. The most common cause of this condition is SIADH. This syndrome is generally associated with diseases of the central nervous system, pulmonary diseases, and neoplasms. These conditions lead to secretion of AVP which is inappropriate both from the standpoint of plasma osmolality and effective arterial volume. A number of other etiologies cause a condition of hypoosmolality associated with euvolemia and can mimic the syndrome of inappropriate AVP secretion. These include isolated glucocorticoid deficiency (normal mineralocorticoid activity), hypothyroidism, pain, nausea, acute psychosis, and a variety of drugs.
Clinical Conditions Associated With Hyponatremia in the Hospital Setting
Post‐Operative Hyponatremia
The postoperative patient is particularly prone to developing hyponatremia. AVP levels are increased for several days following surgical procedures due to baroreceptor and nonbaroreceptor‐mediated mechanisms. These patients typically have subtle or overt decreases in effective arterial blood volume due to prolonged preoperative fasting combined with intraoperative and postoperative blood loss and third spacing of fluid. In addition to these factors which unload baroreceptors, postoperative pain, stress, anxiety, nausea, and administration of morphine can further stimulate the release of AVP. In some instances nonsteroidal antiinflmmatory drugs are given which have the effect of augmenting the hydroosmotic actions of AVP.6 In this setting of compromised ability to excrete a water load, administration of hypotonic fluid can precipitate acute iatrogenic hyponatremia.
Postoperative hyponatremia has been a major problem in pediatric populations due to the widespread practice of using hypotonic fluids for maintenance therapy. This approach is based on guidelines developed 50 years ago which were based on calculations linking energy expenditures to water and electrolyte losses.7 More recently, several groups have argued that isotonic rather than hypotonic fluids should be the routine maintenance fluid in such patients.8, 9 The pediatric community has been slow to embrace this approach out of the concern that excessive administration of Na+ would increase the risk of hypernatremia. A systematic meta‐analysis of studies comparing isotonic and hypotonic fluids in hospitalized children found the odds of developing hyponatremia following hypotonic solutions was 17.2 times greater than with isotonic fluids.10 The concern that isotonic maintenance fluids carry a risk of hypernatremia was not supported in the review. Some studies actually reported a decrease in serum Na+ concentration, presumably due to the desalination phenomenon in which hypertonic urine is excreted in volume expanded subjects with persistent vasopressin secretion.
Endocrine Disorders
Glucocorticoid Deficiency
Patients with glucocorticoid deficiency develop hyponatremia. It is important to separate this condition from that of mineralocorticoid deficiency and combined mineralocorticoid‐glucocorticoid deficiency. In patients with mineralocorticoid deficiency, ECF volume and effective arterial volume are low. This leads to baroreceptor stimulation of AVP secretion and to decreased distal delivery of filtrate to the diluting segments of the nephron. In isolated glucocorticoid deficiency, the patients are euvolemic. In these patients, for any given level of low plasma osmolality, vasopressin levels are inappropriately elevated. The administration of hydrocortisone restores the relationship between vasopressin and plasma osmolality to normal.
While it is possible to develop isolated glucocorticoid deficiency with adrenal disease, most adrenal diseases cause loss of mineralocorticoid and glucocorticoid function. Glucocorticoid deficiency in the absence of mineralocorticoid deficiency is usually due to pituitary disease. In fact, severe hyponatremia may be the initial clue to the presence of previously unrecognized hypopituitarism. Insufficient adrenal secretion of glucocorticoids may also be a complication following the long term use of exogenous glucocorticoids.
In addition to high AVP levels, AVP‐independent mechanisms lead to increases in urinary osmolality in glucocorticoid deficiency. The nature of this AVP‐independent effect is likely multifactorial. First, glucocorticoid deficiency is associated with a diminished cardiac output and an impaired systemic vascular response to hypotension. These changes will lead to a slight decline in glomerular filtration rate and increased volume absorption in the proximal tubule and thin descending limb. As a result, distal delivery of filtrate to the diluting segment will be abnormally low in glucocorticoid deficiency. Second, mineralocorticoid and glucocorticoid deficiency have both been shown to result in increased expression of aquaporin 2 water channels in the collecting duct.11 This later effect will further limit maximal urinary dilution and contribute to net water retention.
These AVP‐independent mechanisms are consistent with the clinical observation that patients with diabetes insipidus appear to improve clinically when they develop coexistent anterior pituitary insufficiency, and treatment of these patients with glucocorticoids appears to worsen the diabetes insipidus. Thus, in the patient with diabetes, insipidus‐free water excretion will be extremely large. When simultaneous glucocorticoid deficiency develops, free‐water excretion decreases.
Hypothyroidism
Myxedema coma is the most severe form of hypothyroidism and is commonly associated with hyponatremia.12 In this setting, blood pressure can be low because of decreased intravascular volume and cardiovascular collapse. The hypotension can be refractory to vasopressor therapy in the absence of thyroid hormone therapy. Cardiac output and stroke volume are low. A defect in renal water excretion develops as a result of baroreceptor mediated increases in AVP along with decreased delivery of filtrate to the distal nephron. In hypothyroid rats, there is upregulation of aquaporin 2 water channels. In these animals, administration of a V2 receptor antagonist reverses the increased water channel expression and corrects the impaired response to an acute water load.11
While reduced cardiac output and blood pressure associated with severe hypothyroidism can provide a stimulatory effect for AVP release through a baroreceptor mediated mechanism, milder forms of hypothyroidism can be considered in the differential diagnosis of euvolemic hyponatremia. In this setting, impaired renal excretion of water is presumably due to increased release of AVP due to the absence of a tonic inhibitory effect of thyroid hormone in the central nervous system. The degree of hyponatremia in this setting is typically mild.
Heart Failure
Hyponatremia is a common complication of left‐sided heart failure and several studies have shown that it is an independent predictor of mortality.13, 14 A similar association between reduced survival and hyponatremia is present in advanced right‐sided heart failure in patients with pulmonary arterial hypertension.15 Patients with heart failure who are hyponatremic have higher circulating levels of neurohormones (catecholamines, renin, angiotensin II, aldosterone, and AVP) than normonatremic subjects and are more likely to have prerenal azotemia. In addition to being a marker for the extent of neurohumoral activation, hyponatremia may play a more direct role in adverse outcomes through maladaptive volume regulatory responses of cardiac myocytes and by direct effects of AVP on cardiac and coronary V1a receptors.
Heart failure is associated with arterial underfilling leading to arterial baroreceptor‐mediated activation of the neurohumoral axis. This underfilling is due to decreased cardiac output in low‐output heart failure and decreased systemic vascular resistance in high‐output heart failure. Activation of the sympathetic nervous system along with the renin‐angiotensin‐aldosterone system leads to renal salt retention while the increase in AVP is associated with water retention and hyponatremia.
Cirrhosis
Hyponatremia is a common electrolyte abnormality in patients with cirrhosis and occurs with a frequency that tends to parallel the severity of liver disease.16 Patients with a serum Na+ 130 mEq/L are more likely to have refractory ascites and require therapeutic paracentesis. Hepatic encephalopathy, hepatorenal syndrome, and spontaneous bacterial peritonitis are also more common in patients with a serum Na+ 130 mmol/L than in patients with a normal serum Na+ concentration. Hyponatremia increases morbidity and mortality from hepatic transplantation and is associated with osmotic demyelination in the postoperative period because of the large increase in Na+ concentration associated with the procedure.17
Hepatic cirrhosis is characterized by a decreased effective arterial blood volume and activation of neurohumoral effectors. Reduced effective circulating volume due to generalized and specifically to arterial splanchnic vasodilation leads to baroreceptor‐mediated nonosmotic stimulation of AVP release and an impaired ability to excrete electrolyte‐free water.18 Reduced Na+ delivery to the distal tubule because of a low glomerular filtration rate and increased proximal Na+ reabsorption adds to the susceptibility of cirrhotic patients to hyponatremia.
Hyponatremic‐Hypertensive Syndrome
The development of hyponatremia in patients with severe hypertension associated with renal artery stenosis has been called the hyponatremic‐hypertensive syndrome.19, 20 Patients with this syndrome present with a variety of signs and symptoms that include headache, confusion, postural dizziness, polyuria, polydipsia, and salt craving. In a retrospective review of 32 patients with this syndrome, most of the subjects were thin, elderly, women smokers who had atherosclerotic renal vascular disease.19 Biochemical abnormalities included not only hyponatremia, but hypokalemia and increased plasma renin activity. The mean serum Na+ concentration was 129.7 mmol/L (range 120‐135).
The precise mechanism of this syndrome is not known. Given the available data, angiotensin‐mediated thirst coupled with nonosmotic release of AVP provoked by angiotensin II and/or hypertensive encephalopathy are likely. Na+ depletion due to pressure natriuresis, and K+ depletion due to hyperaldosteronism are also likely to play a role in the pathogenesis of hyponatremia.
Pneumonia
An association between pneumonia and hyponatremia has been known for quite some time but has been poorly defined. Of the various etiologic agents, legionella is more commonly associated with hyponatremia as compared to other types of community acquired disease. As has been true for many disorders, hyponatremia is associated with longer hospital stays and hospital mortality, most likely reflecting the severity of the pneumonia rather than morbidity from the usually mild and asymptomatic hyponatremia.
Central Nervous System Disease
Hyponatremia is a frequent complication of central nervous system disease to include bacterial meningitis and traumatic brain injury. Potential mechanisms include the development of SIADH, cerebral salt wasting (CSW), or hypotonic fluid administration in the setting of impaired renal water excretion as in patients with low effective volume. Of these various causes, hyponatremia is frequently attributed to SIADH. As previously mentioned, this syndrome is characterized by hyponatremia in the setting of an inappropriately concentrated urine, increased urine Na+ concentration, and evidence of normal or slightly increased intravascular volume.
However there are patients with intracranial disease who develop hyponatremia with similar characteristics but differ in that there is clinical evidence of a contracted ECF volume. This form of hyponatremia is due to excessive renal Na+ excretion resulting from a centrally mediated process and is termed CSW. The onset of this disorder is typically seen within the first ten days following a neurosurgical procedure or after a definable event, such as a subarachnoid hemorrhage or stroke. CSW has also been described in other intracranial disorders, such as carcinomatous or infectious meningitis and metastatic carcinoma.21 The distinction between SIADH and CSW is of considerable clinical importance given the divergent nature of the treatments. Fluid restriction is the treatment of choice in SIADH, whereas the treatment of CSW comprises vigorous Na+ and volume replacement.
Treatment of Hyponatremia
Symptoms of hyponatremia include nausea and malaise, which can be followed by headache, lethargy, muscle cramps, disorientation, restlessness and obtundation, and seizures. The principal danger of hyponatremia or hypernatremia relates to effects on central nervous system function due to changes in brain size (Figure 2).
Hyponatremia initially leads to cell swelling driven by the higher intracellular osmolality. The net result is equilibration of intracellular and extracellular osmolality at the expense of increased brain volume. Cells in general, and brain cells in particular, then respond by decreasing the number of intracellular osmoles and as intracellular osmolality decreases, cell size returns toward normal despite the presence of hyponatremia. If the decrease in ECF osmolality is slow, there will be no measurable cell swelling. This pathophysiologic sequence correlates well with clinical observations. If hyponatremia is slow in onset, neurologic symptoms and permanent brain damage are unusual, even if the decreases in Na+ concentration and ECF osmolality are large. Conversely, if hyponatremia is rapid in onset, cerebral edema and significant CNS symptoms and signs can occur with lesser changes in serum Na+ concentration.
When treating a patient with hyponatremia, the Na+ concentration should be raised at the rate at which it fell. In a patient whose serum Na+ concentration has decreased rapidly (<48 hours), neurologic symptoms are frequently present and there is cerebral edema. In this setting there has not been sufficient time to remove osmoles from the brain and rapid return to normal ECF osmolality merely returns brain size to normal. In general, the development of hyponatremia in the outpatient setting is more commonly chronic in duration and should be corrected slowly. By contrast, hyponatremia of short duration is more likely to be encountered in hospitalized patients receiving intravenous free water. Use of ecstasy, exercise‐induced hyponatremia, or patients with primary polydipsia can also develop acute hyponatremia and if symptomatic may similarly require rapid correction. Raising the serum Na+ concentration by 4 mEq/L to 6 mEq/L over a several hour period is both safe and effective in preventing untoward effects of acute hyponatremia.22 A reasonable strategy to accomplish this goal is the regimen recommended for the treatment of athletes with hyponatremia and encephalopathy.23 These guidelines suggest an immediate bolus of 100 mL 3% NaCl (513 mEq/L). If there is no neurologic improvement two additional 100 mL 3% NaCl bolus infusions separated by 10 minute intervals can be given.
In patients with chronic hyponatremia (>48 hours duration) the serum Na+ concentration has fallen slowly. Neurologic symptoms are generally minimal, brain size is normal, and the number of intracellular osmoles is decreased. Sudden return of ECF osmolality to normal values will lead to cell shrinkage and possibly precipitate osmotic demyelination. Experts in the field suggest this complication can be prevented by adhering to the following limits of correction: 10 mmol/L in 24 hours, 18 mmol/L in 48 hours, and 20 mmol/L in 72 hours.22 In order to maximize patient safety, the goals of therapy should be more modest: 6 to 8 mmol/L in 24 hours, 12 to 14 mmol/L in 48 hours, and 14 to 16 mmol/L in 72 hours.
A formula designed to predict the increase in serum Na+ concentration to be expected from the infusion of 1 L of a given infusion is given below:
| Treament | Mechanism | Advantages | Limitations |
|---|---|---|---|
| |||
| Fluid restriction (0.5‐1.0 L/day) | water intake | Effective, inexpensive | Poor compliance |
| Demeclocyline (600‐1200 mg/day) | Inhibits action of AVP | Widely available | Slow onset, nephrotoxic, expensive |
| Urea (30 gm/day) | Osmotic diuresis | Possiblerisk of osmotic demyelination | Poor palatability, avoid in chronic kidney and liver disease |
| Lithium (up to 900 mg/daily) | Inhibits action of AVP | Widely available | Slow onset, toxicity |
When treating patients with SIADH with intravenous salt solutions, one can worsen the degree of hyponatremia if the osmolality of the administered fluid is less that the urine osmolality. One liter of isotonic saline contains 150 mEq each of Na+ and Cl and has an osmolality of 300 mOsm. In a patient with SIADH who has a urine osmolality of 600 mOsm, the administered NaCl will all be excreted in 500 mL of water (300 mOsm in 500 mL = 600 mOsm/L). The remaining 500 mL of administered fluid will be retained in the body and cause further dilution of the serum Na+ concentration. By contrast, 1 L of 3% saline contains 513 mEq each of Na+ and Cl and has an osmolality of 1026 mOsm. All of the NaCl in 1 L of this solution will be excreted in 1.7 L of water and therefore will cause the serum Na+ concentration to increase as a result of the loss of 700 mL of water.
Loop diuretics can also be employed as part of the strategy to treat chronic hyponatremia. Loop diuretics directly interfere in the countercurrent concentrating mechanism by inhibiting NaCl reabsorption in the loop of Henle. The coadministration of these drugs will enhance the effect of intravenous salt solutions by lowering urine osmolality thereby increasing the likelihood that the osmolality of the administered fluid is greater than the urine osmolality. Other measures used to treat chronic hyponatremia are given in Table 1.
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of water channels (aquaporin 2) into the apical membrane. The net effect is increased water excretion with little to no change in urinary Na+ excretion. AVP antagonists have been shown to be more effective than placebo in increasing the serum Na+ concentration in patients with modest asymptomatic hyponatremia.26, 27 These agents can be considered in patients with hyponatremia associated with heart failure or cirrhosis and in patients with irreversible SIADH but should not be used in patients with hypovolemic hyponatremia. The role of these drugs is discussed further in an accompanying article in this supplement, titled New horizons in the pharmacologic approach to hyponatremia: The V2 Receptor Antagonists.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chem Acta.2003;337:169–172.
- ,,,.Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin.Ann Intern Med.1985;102(2):164–168.
- ,,, et al.Characteristics and mortality of severe hyponatraemia‐a hospital‐based study.Clin Endocrinol (Oxf).2006;65(2):246–249.
- ,,.Mortality after hospitalization with mild moderate and severe hyponatremia.Am J Med.2009;122:8857–8865.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- .Renal complications associated with use of nonsteroidal anti‐inflammatory agents.J Investig Med.1995;43(6):516–533.
- ,.Recent developments in the perioperative fluid management for the paediatric patient.Curr Opin Anaesthesiol.2006;19:268–277.
- ,.Prevention of hospital‐acquired hyponatremia: a case for using isotonic saline.Pediatrics.2003;111:227–230.
- ,,,,.Acute hyponatremia related to intravenous fluid administration in hospitalized children: an observational study.Pediatrics.2004;113(5):1279–1284.
- ,,,.Hypotonic versus isotonic saline in hospitalized children: a systematic review.Arch Dis Child.2006;91(10):828–835.
- .Vasopressin and Aquaporin 2 in clinical disorders of water homeostasis.Semin Nephrol.2008;28:289–296.
- .Myxedema coma.Edocrinol Metab Clin N Am.2006;35:687–698.
- ,,, et al.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28(8):920–921.
- ,.Pulmonary hypertension, right ventricular failure, and kidney: different from left ventricular failure.Clin J Am Soc Nephrol.2008;3:1232–1237.
- ,,, et al.Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension.Am J Respir Crit Care Med.2008;177(12):1364–1369.
- ,,,;CAPPS investigators. Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,,,,.Possible causes of central pontine myelinolysis after liver transplantation.World J Gastroenterol.2004;10(17):2540–2543.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.The hyponatremic hypertensive syndrome in renal artery stenosis: An infrequent cause of hyponatremia.J Postgrad Med.2007;53:41–43.
- ,,,.Hyponatremic‐hypertensive syndrome with renal ischemia: an underrecognized disorder.Hypertension.1993;33:1020–1024.
- .Hyponatremia in patients with central nervous system disease: SIADH or CSW.Trends Endocrinol Metab.2003;14:182–187.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- ,,, et al.Statement of the second international exercise‐associated hyponatremia consensus development conference, New Zealand.Clin J Sport Med.2008;18:111–121.
- ,,,,,.Hypertonic saline for hyponatremia: risk of inadvertent overcorrection.Clin J Am Soc Nephrol.2007;2:1110–1117.
- ,,, et al.DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia.Clin J Am Soc Nephrol.2008;3:331–336.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol2007;27:447–457.
- ,,, et al.,Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chem Acta.2003;337:169–172.
- ,,,.Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin.Ann Intern Med.1985;102(2):164–168.
- ,,, et al.Characteristics and mortality of severe hyponatraemia‐a hospital‐based study.Clin Endocrinol (Oxf).2006;65(2):246–249.
- ,,.Mortality after hospitalization with mild moderate and severe hyponatremia.Am J Med.2009;122:8857–8865.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- .Renal complications associated with use of nonsteroidal anti‐inflammatory agents.J Investig Med.1995;43(6):516–533.
- ,.Recent developments in the perioperative fluid management for the paediatric patient.Curr Opin Anaesthesiol.2006;19:268–277.
- ,.Prevention of hospital‐acquired hyponatremia: a case for using isotonic saline.Pediatrics.2003;111:227–230.
- ,,,,.Acute hyponatremia related to intravenous fluid administration in hospitalized children: an observational study.Pediatrics.2004;113(5):1279–1284.
- ,,,.Hypotonic versus isotonic saline in hospitalized children: a systematic review.Arch Dis Child.2006;91(10):828–835.
- .Vasopressin and Aquaporin 2 in clinical disorders of water homeostasis.Semin Nephrol.2008;28:289–296.
- .Myxedema coma.Edocrinol Metab Clin N Am.2006;35:687–698.
- ,,, et al.Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: an analysis from the OPTIMIZE‐HF registry.Eur Heart J.2007;28(8):920–921.
- ,.Pulmonary hypertension, right ventricular failure, and kidney: different from left ventricular failure.Clin J Am Soc Nephrol.2008;3:1232–1237.
- ,,, et al.Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension.Am J Respir Crit Care Med.2008;177(12):1364–1369.
- ,,,;CAPPS investigators. Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44(6):1535–1542.
- ,,,,,.Possible causes of central pontine myelinolysis after liver transplantation.World J Gastroenterol.2004;10(17):2540–2543.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.The hyponatremic hypertensive syndrome in renal artery stenosis: An infrequent cause of hyponatremia.J Postgrad Med.2007;53:41–43.
- ,,,.Hyponatremic‐hypertensive syndrome with renal ischemia: an underrecognized disorder.Hypertension.1993;33:1020–1024.
- .Hyponatremia in patients with central nervous system disease: SIADH or CSW.Trends Endocrinol Metab.2003;14:182–187.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- ,,, et al.Statement of the second international exercise‐associated hyponatremia consensus development conference, New Zealand.Clin J Sport Med.2008;18:111–121.
- ,,,,,.Hypertonic saline for hyponatremia: risk of inadvertent overcorrection.Clin J Am Soc Nephrol.2007;2:1110–1117.
- ,,, et al.DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia.Clin J Am Soc Nephrol.2008;3:331–336.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol2007;27:447–457.
- ,,, et al.,Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.