User login
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
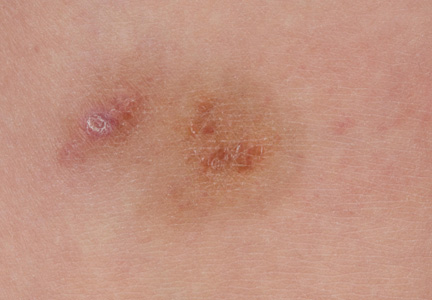
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
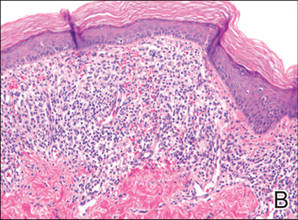
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
 |  |  | ||
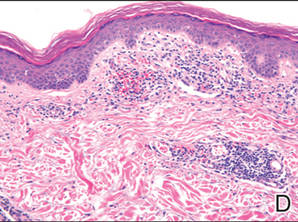 | 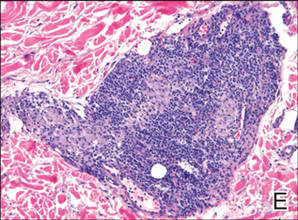 |  | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
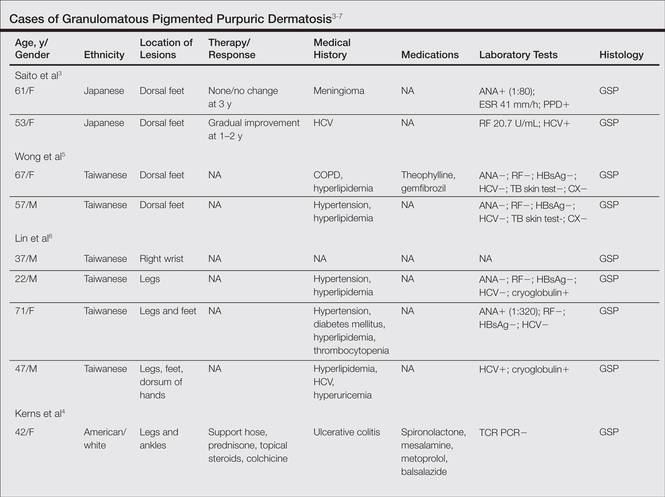
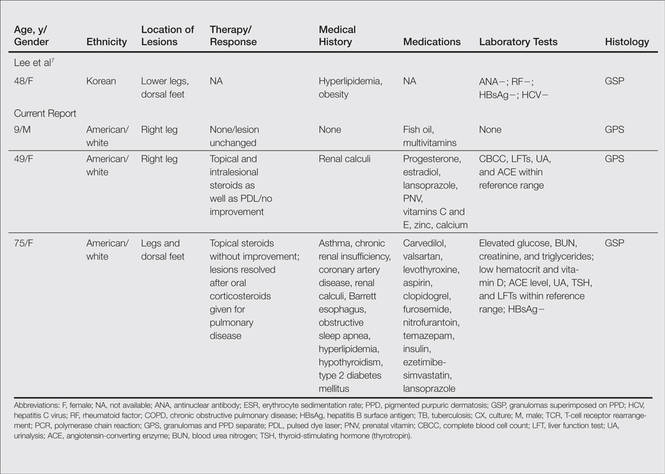
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
| | | ||
| | | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
| | | ||
| | | ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Practice Points
- Consider a punch biopsy when sampling suspected inflammatory dermatoses, such as pigmented purpuric dermatosis, to allow deeper sampling.
- Provide all clinical details to the dermatopathologist to assist with clinicopathologic correlation and diagnostic accuracy.