User login
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).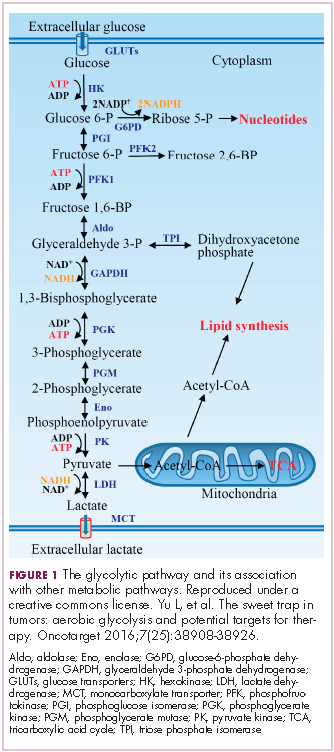
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.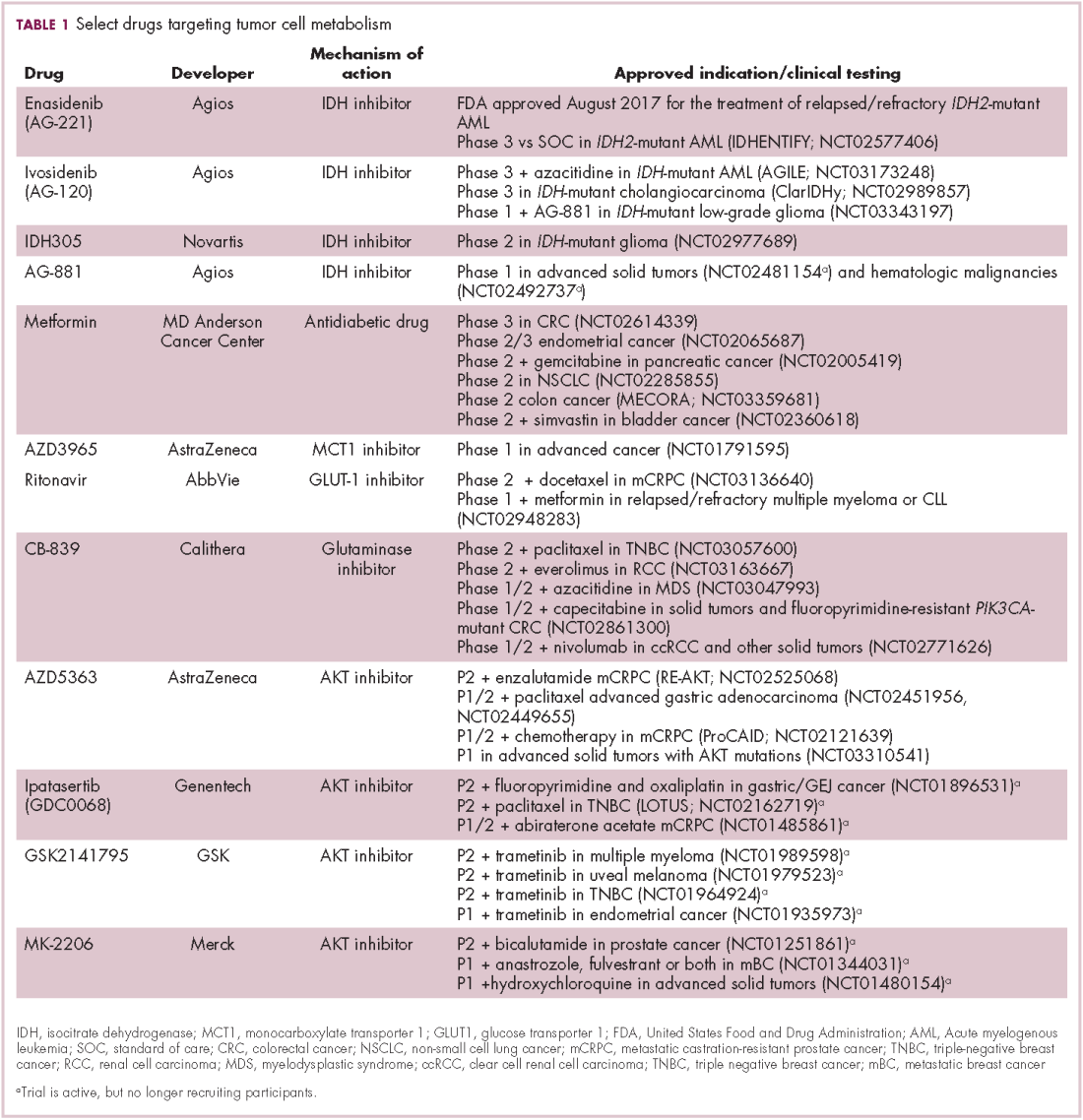
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
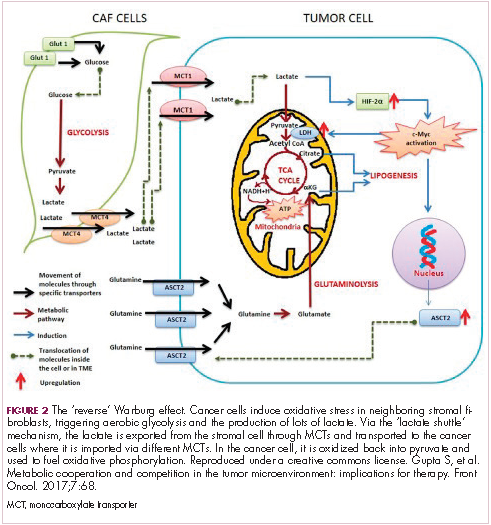
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16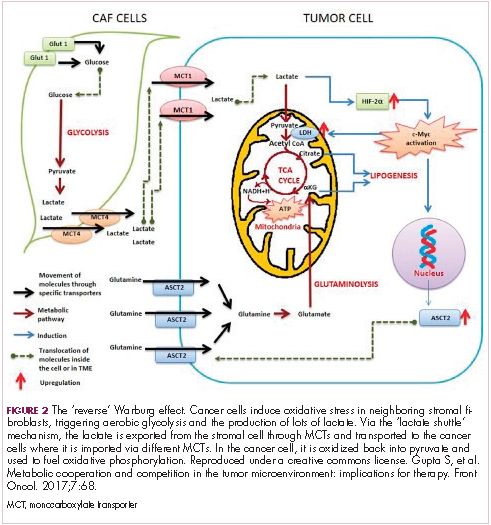
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21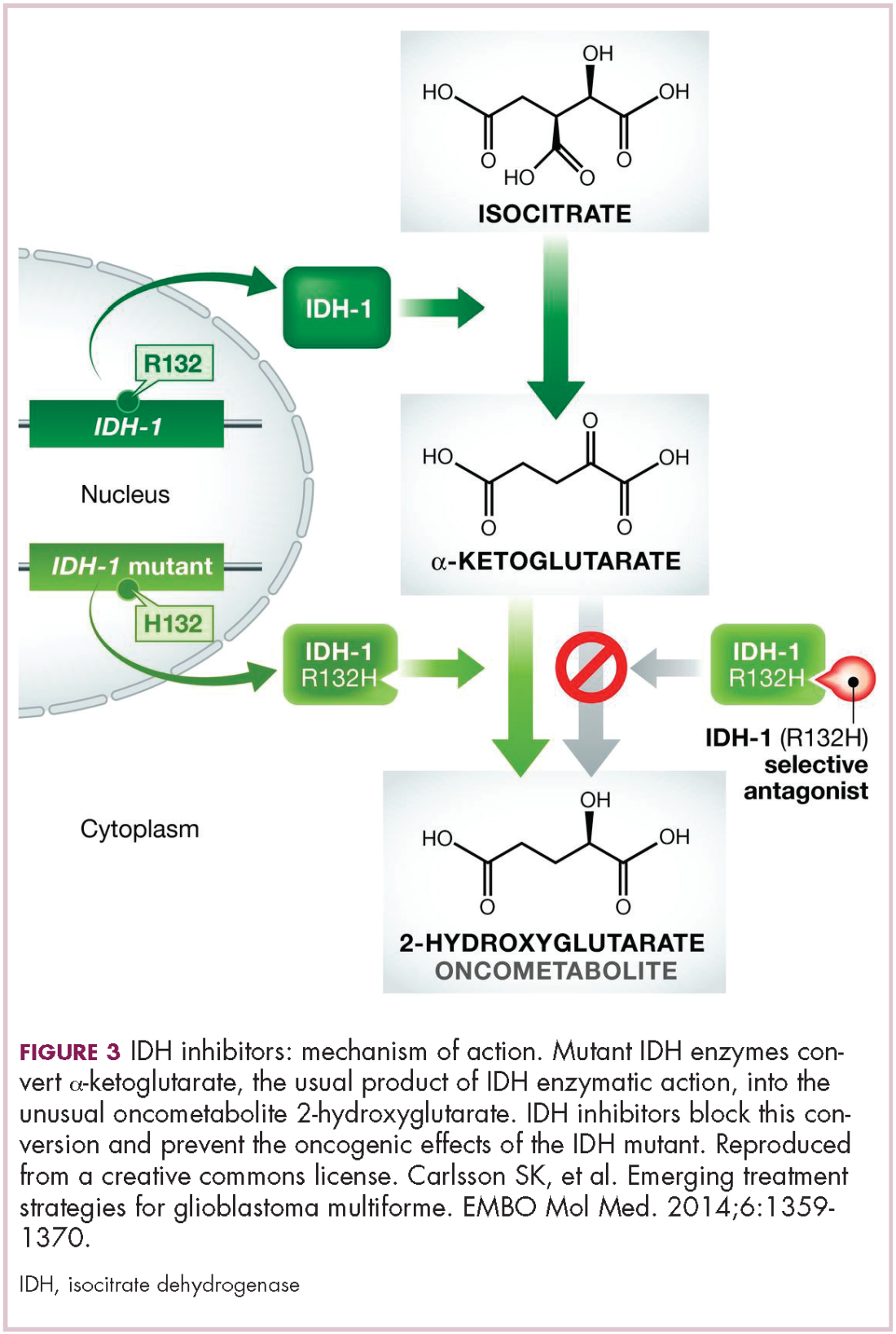
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.