User login
Emerging biosimilars market presents opportunities and challenges
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
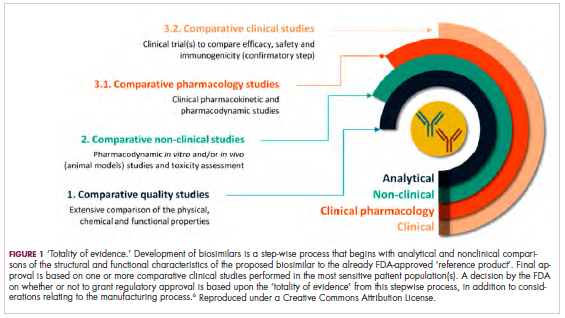
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6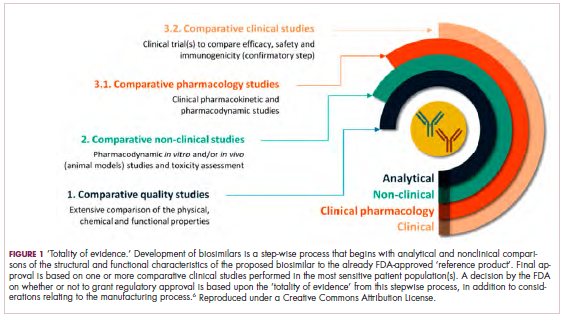
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10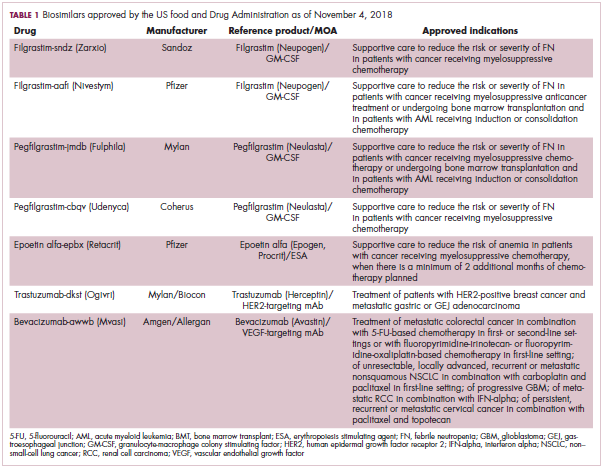
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22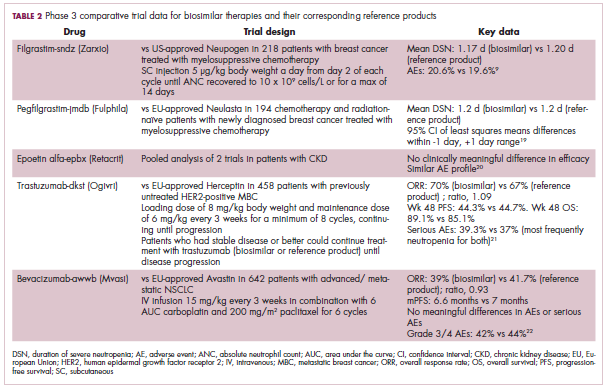
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.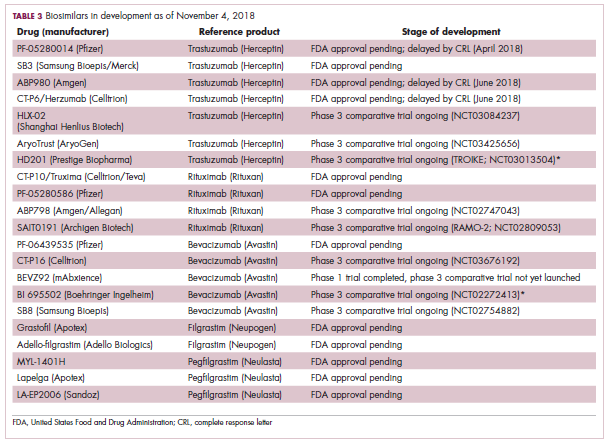
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
Paradigm-changing osimertinib approval in front-line for advanced NSCLC
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.

BRAF-MEK inhibitor combo approved for adjuvant melanoma therapy
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.

Immunotherapy may hold the key to defeating virally associated cancers
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).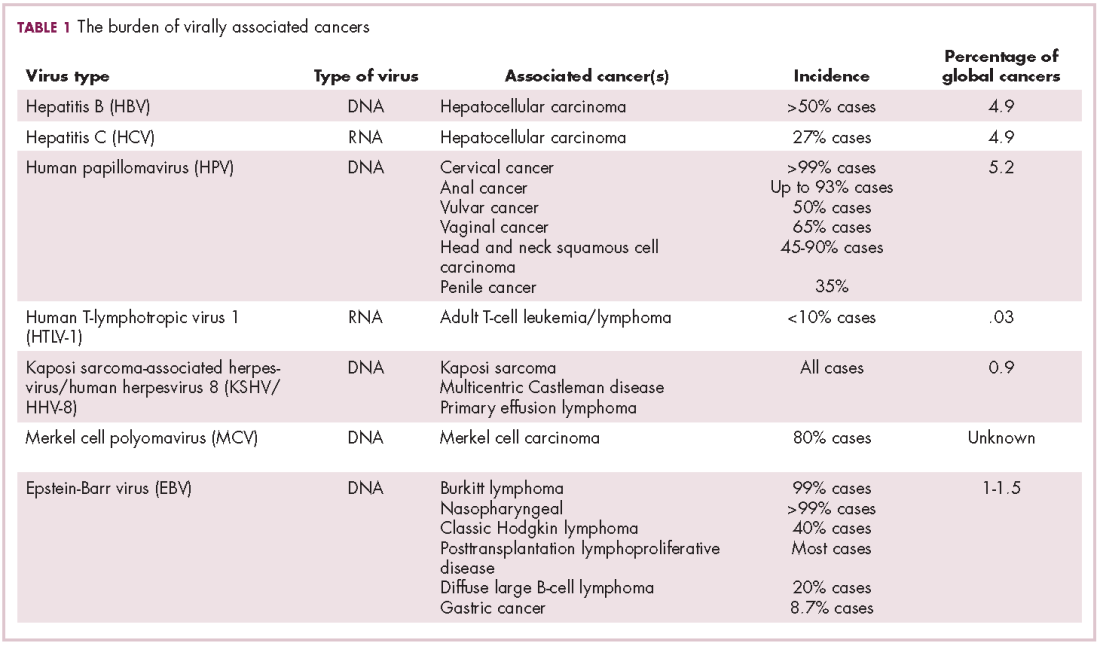
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9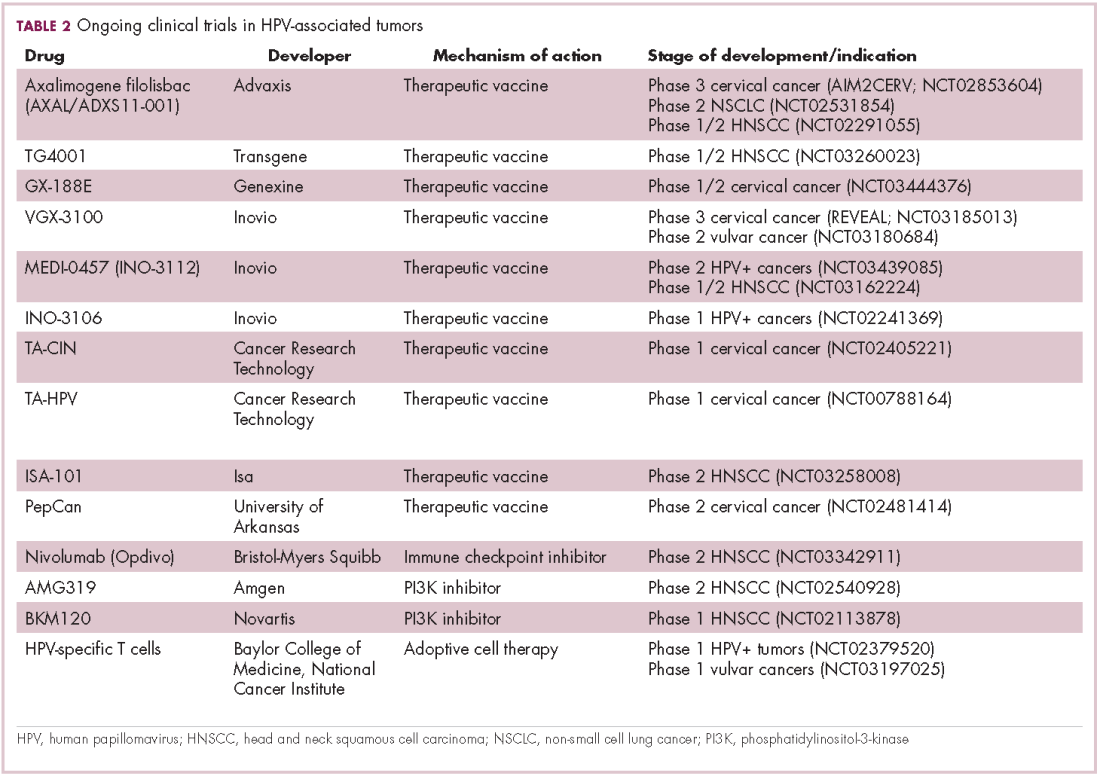
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).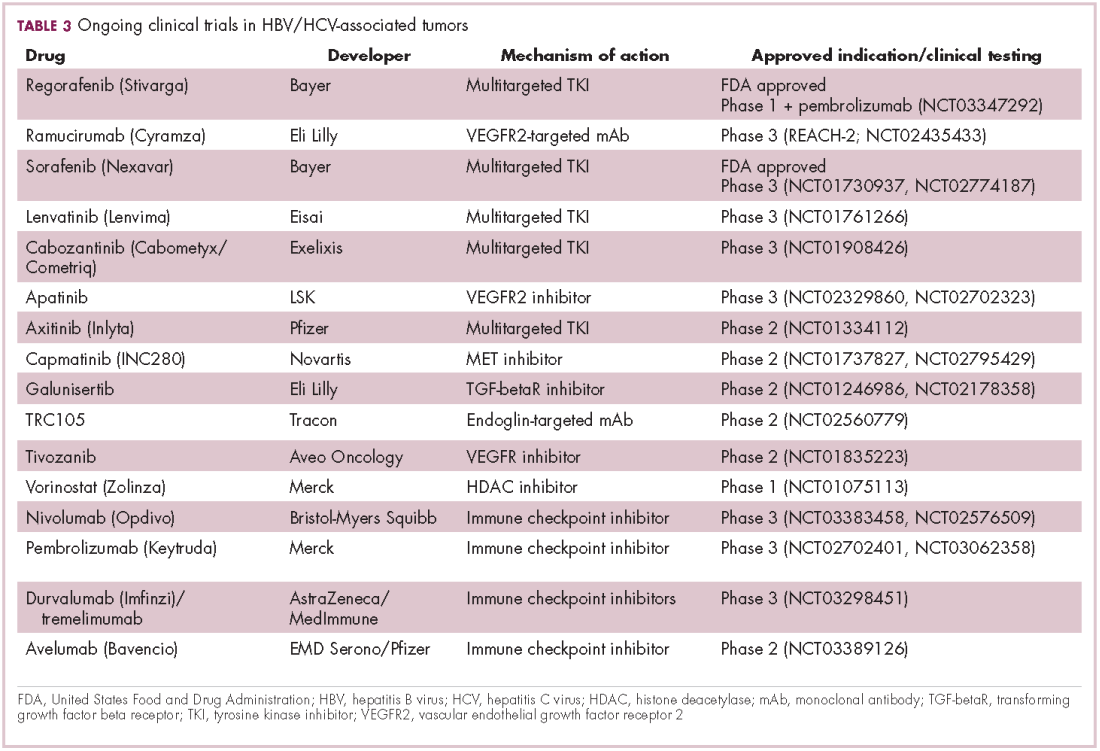
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).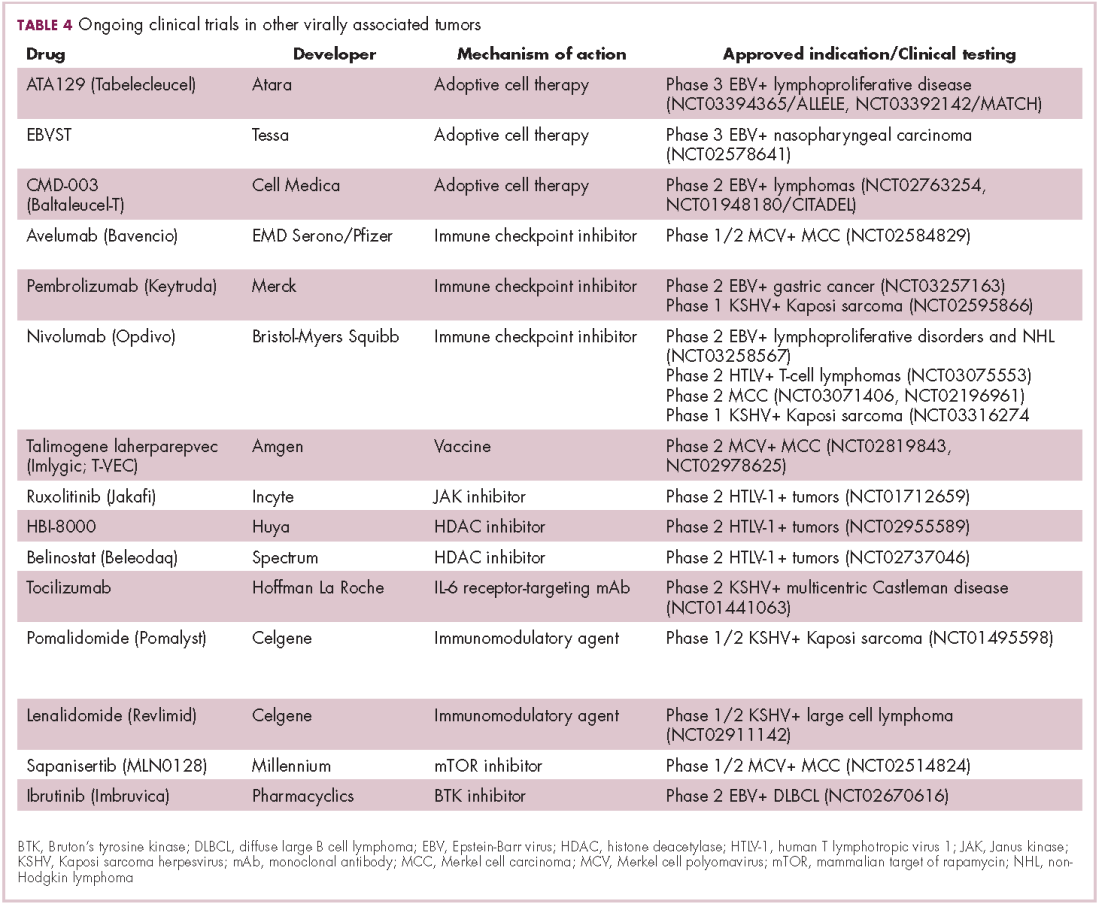
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
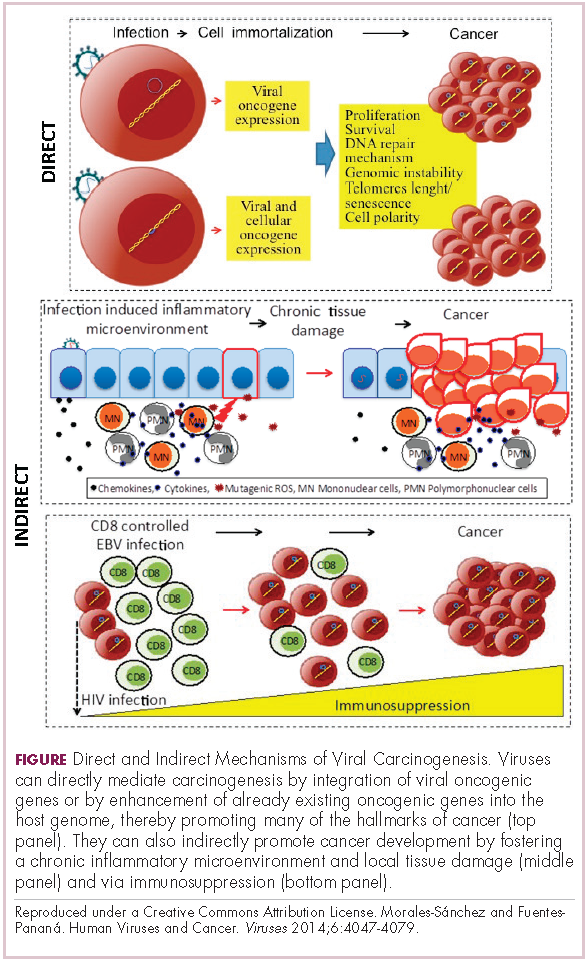
Game changers in pediatric cancer
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
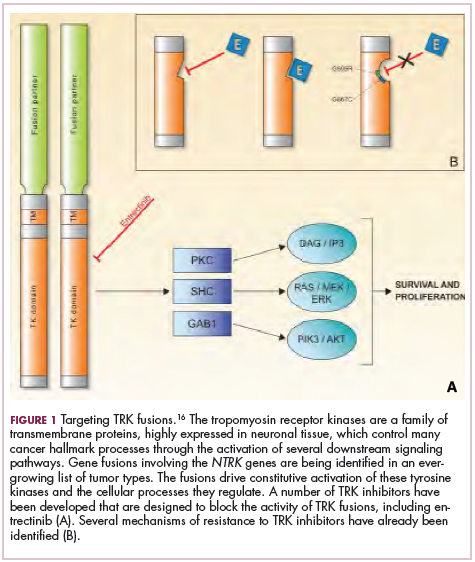
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.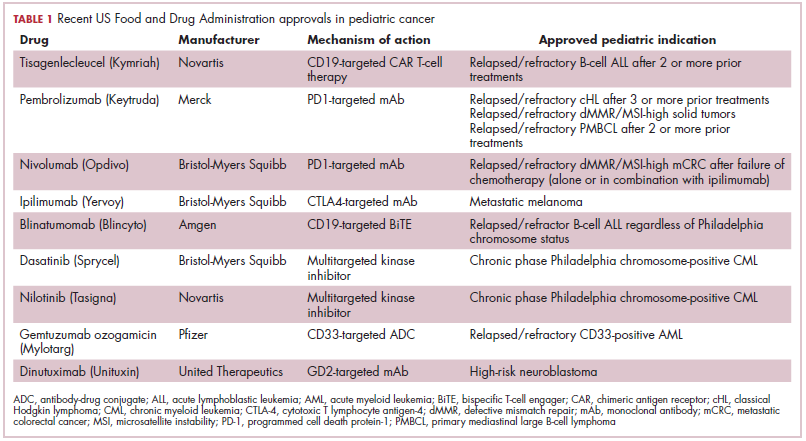
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).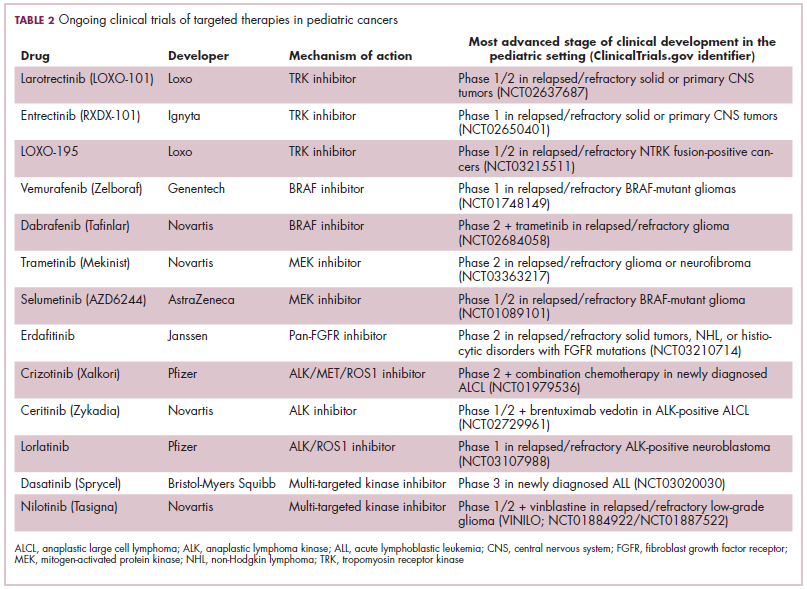
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.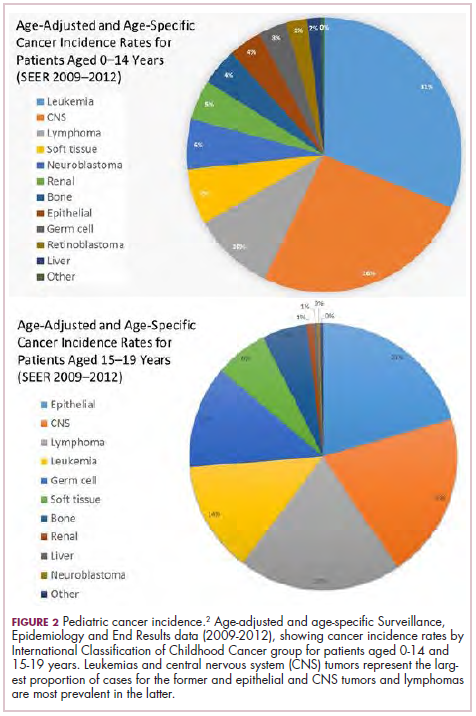
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).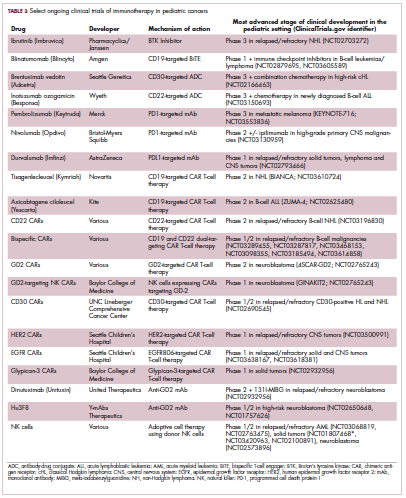
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
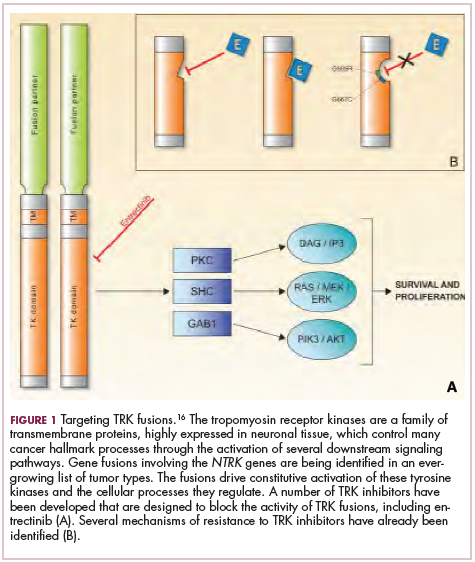
Addressing the rarity and complexities of sarcomas
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3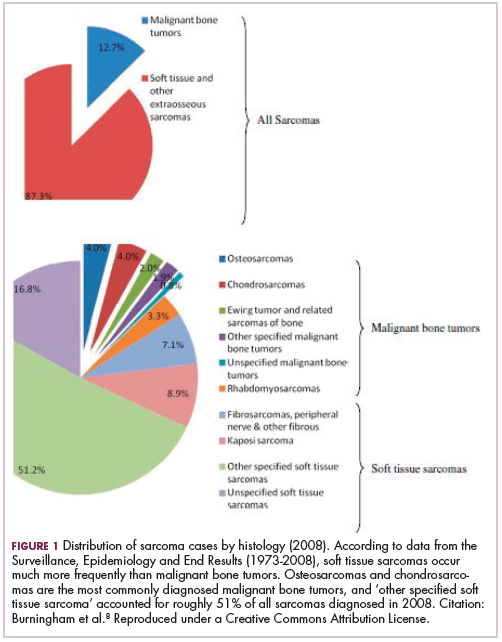
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).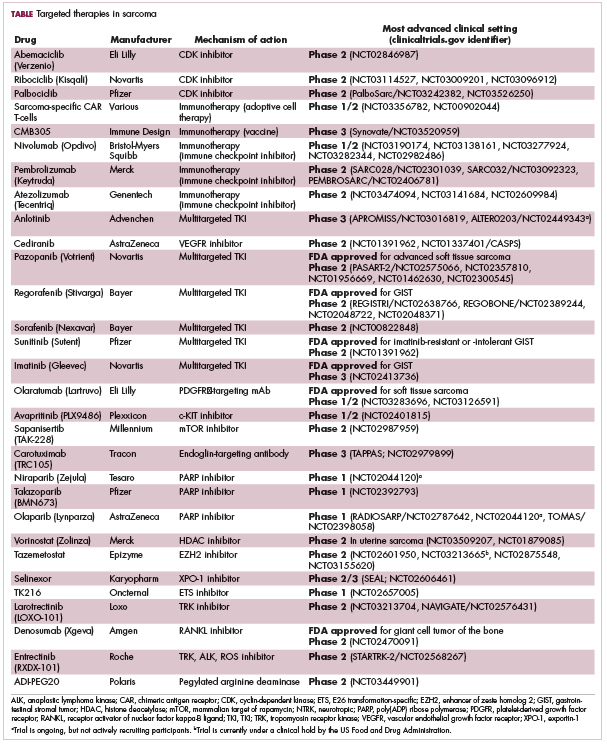
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33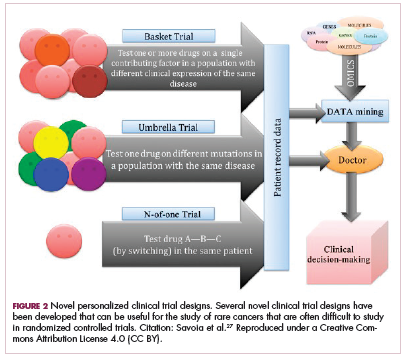
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
Tumor heterogeneity: a central foe in the war on cancer
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.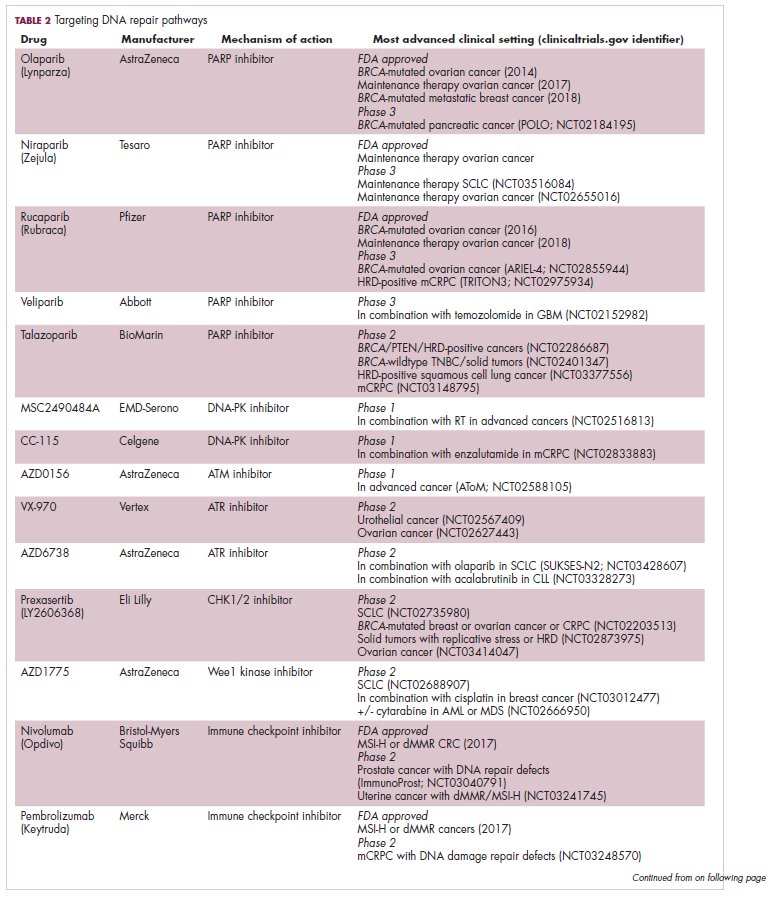

Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).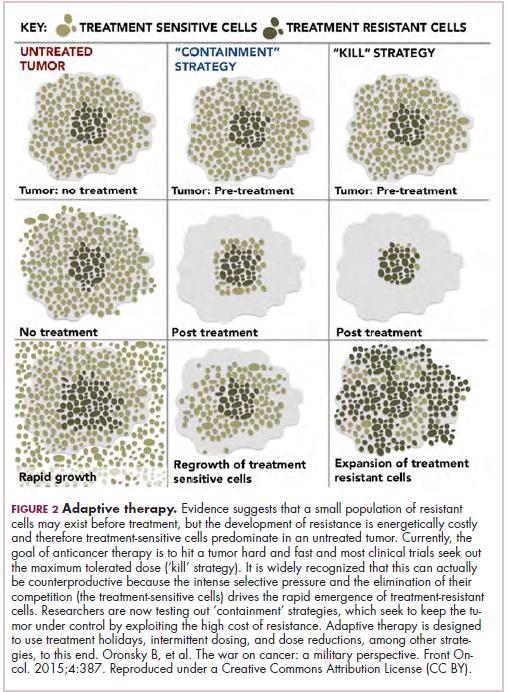
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22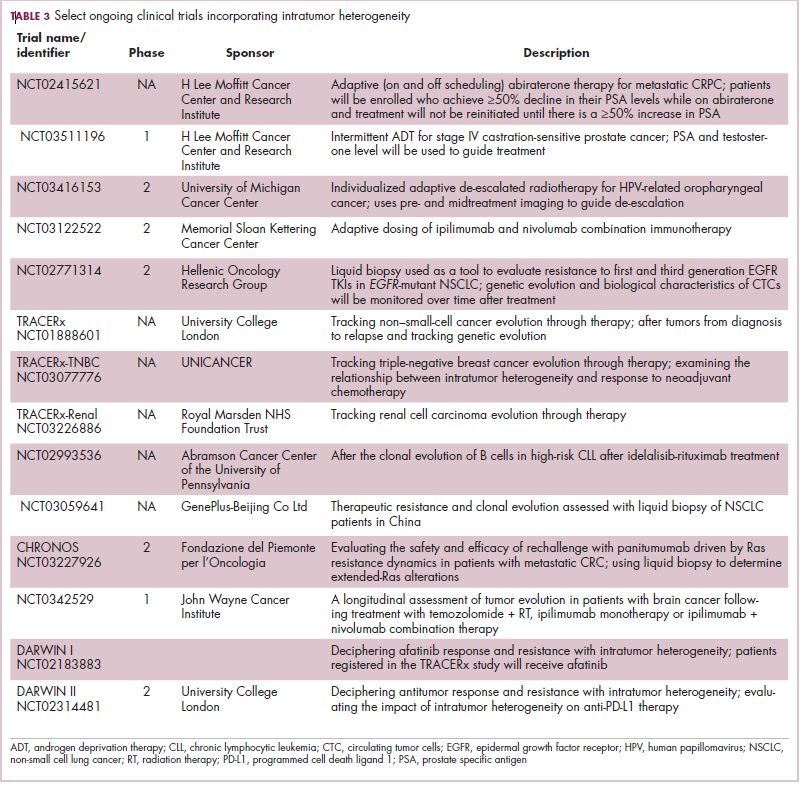
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.
Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.
Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
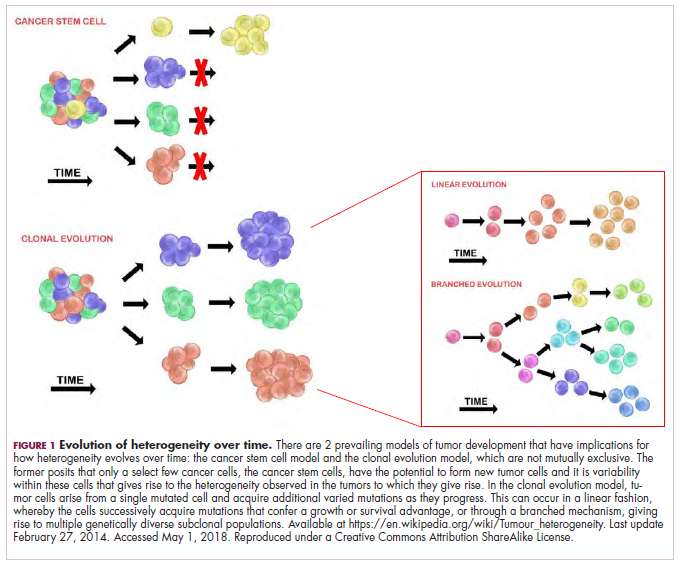
Expanding treatment options for diverse neuroendocrine tumors
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).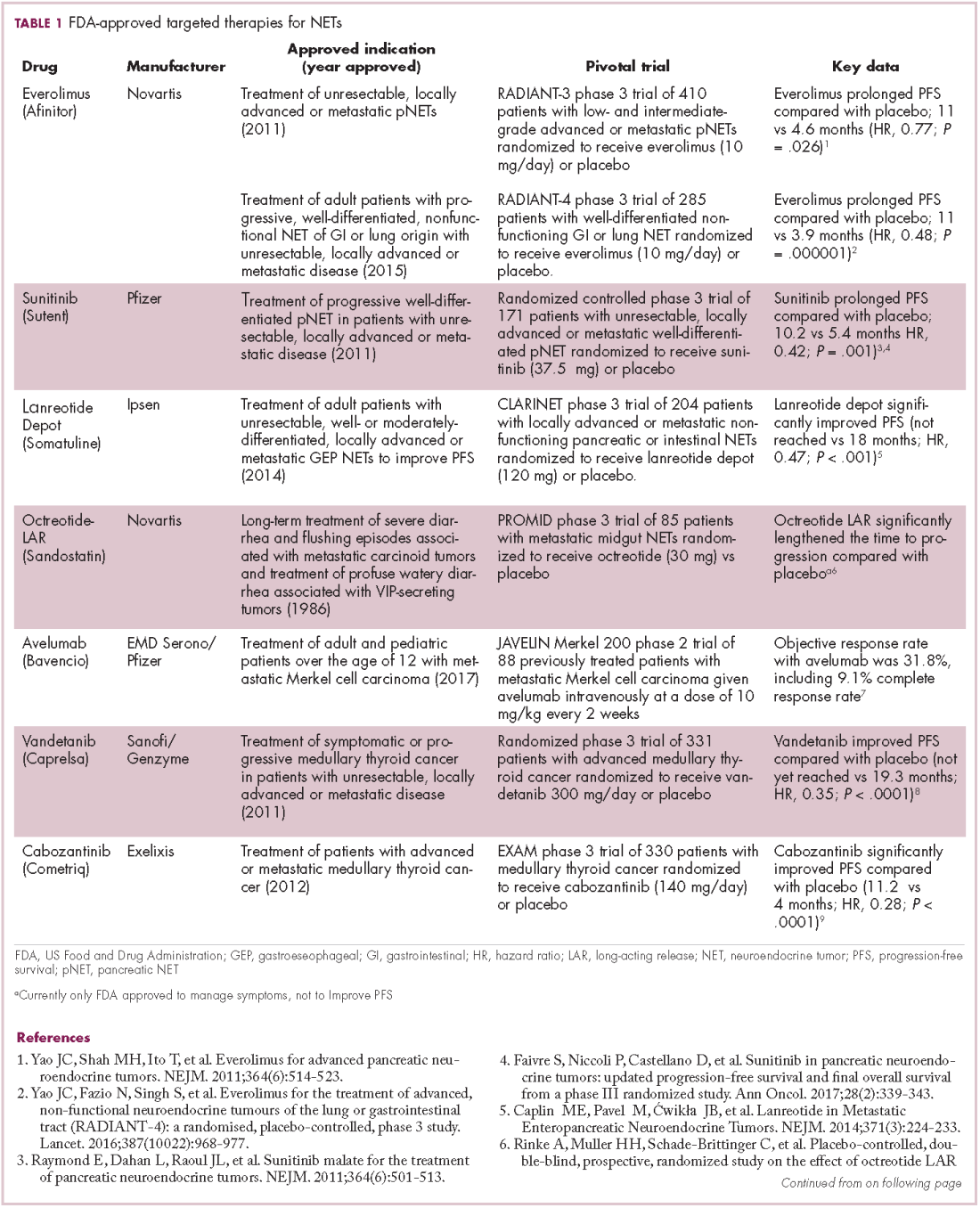

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).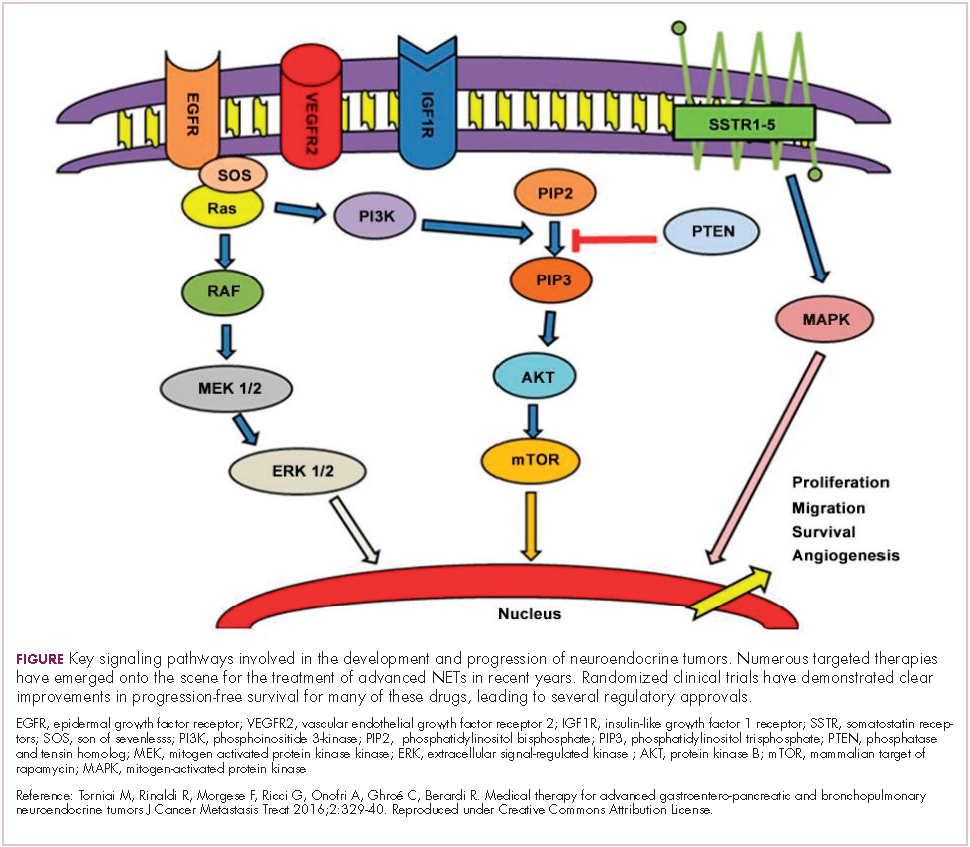
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24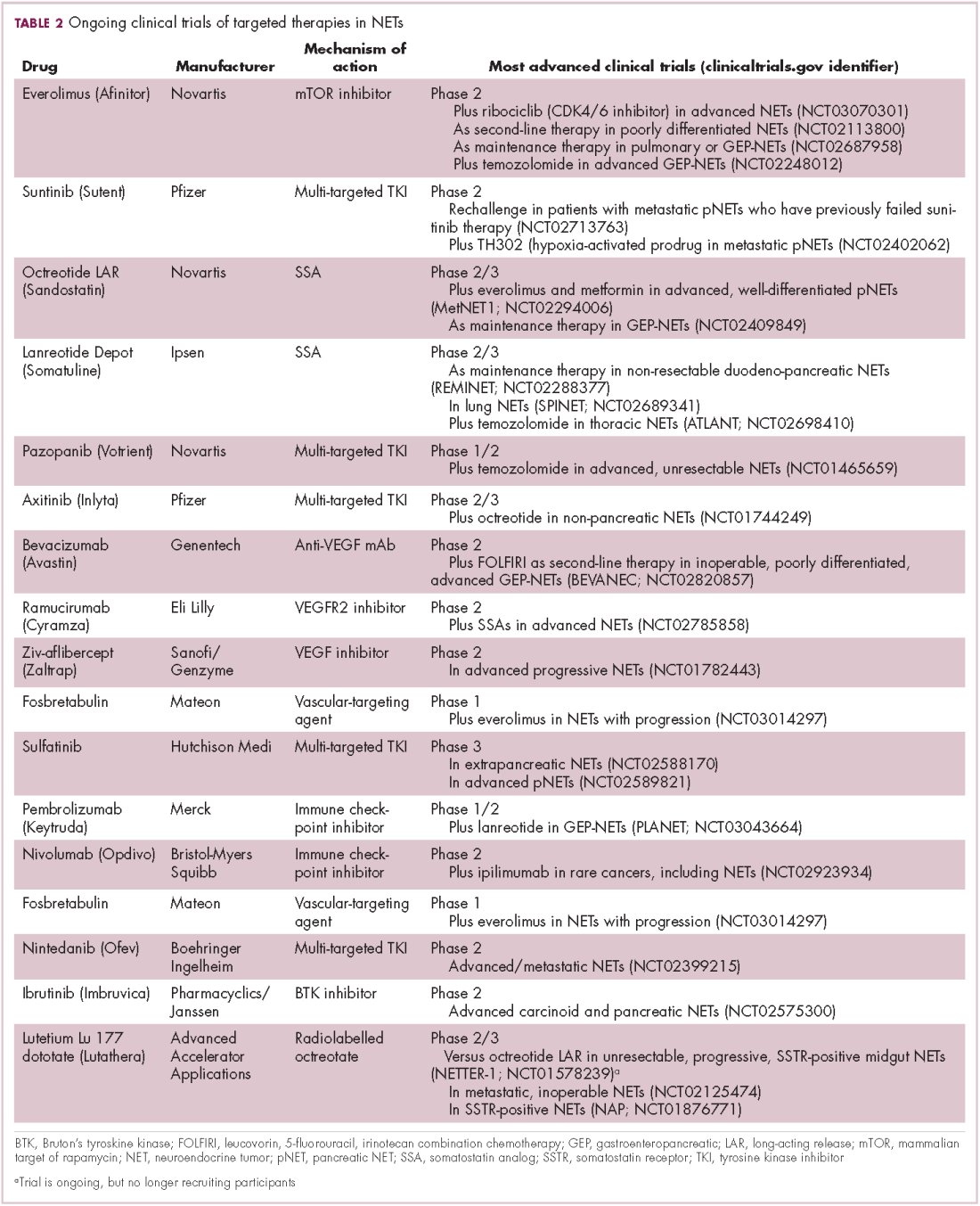
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
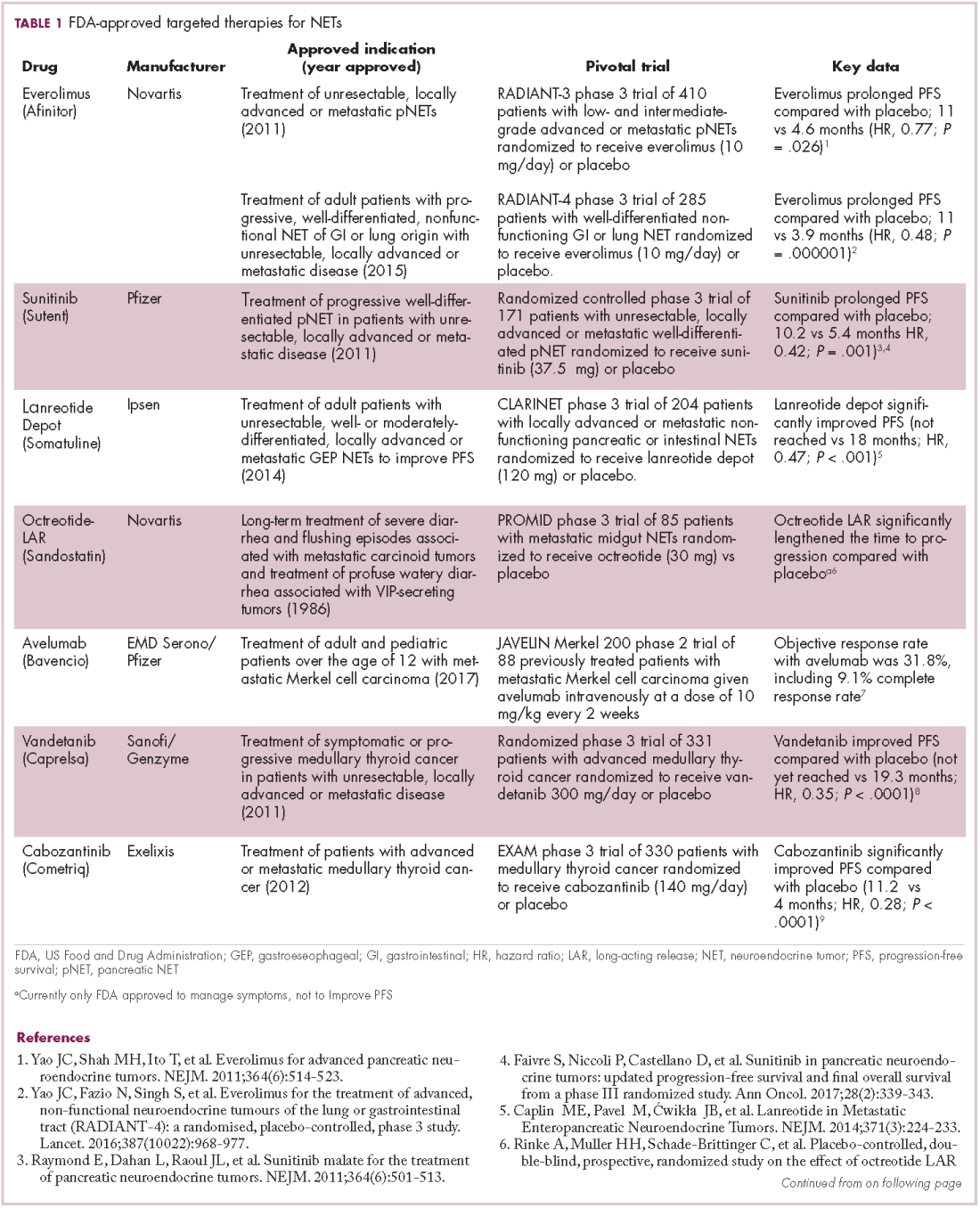
Hallmark tumor metabolism becomes a validated therapeutic target
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).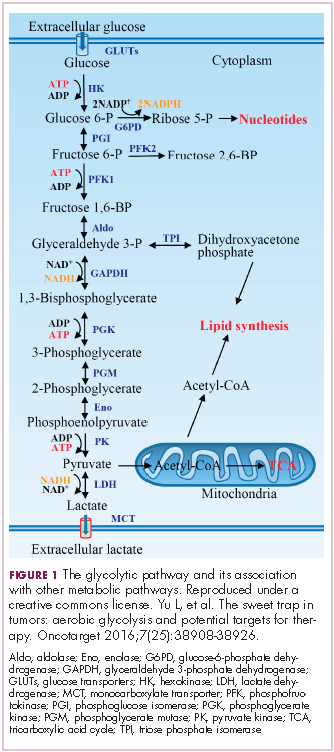
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
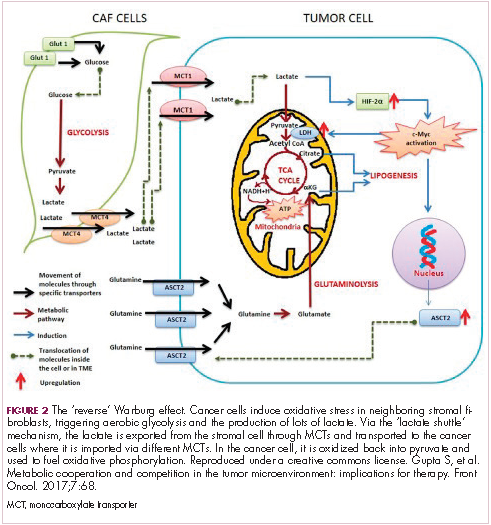
Targeted therapies forge ahead in multiple breast cancer subtypes
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1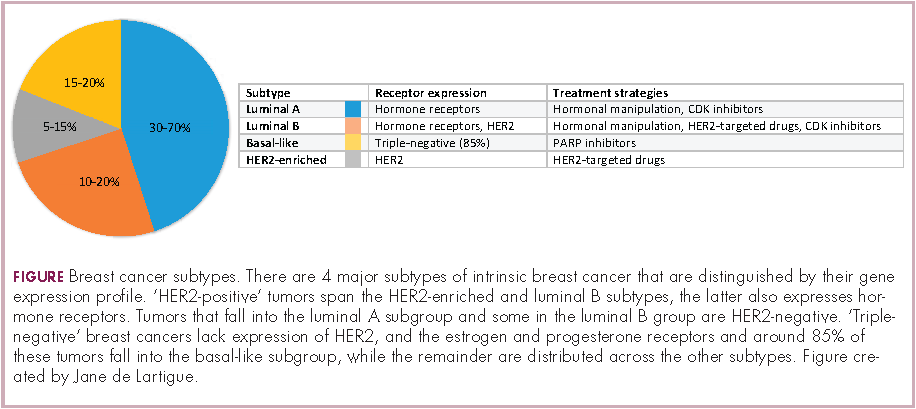
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).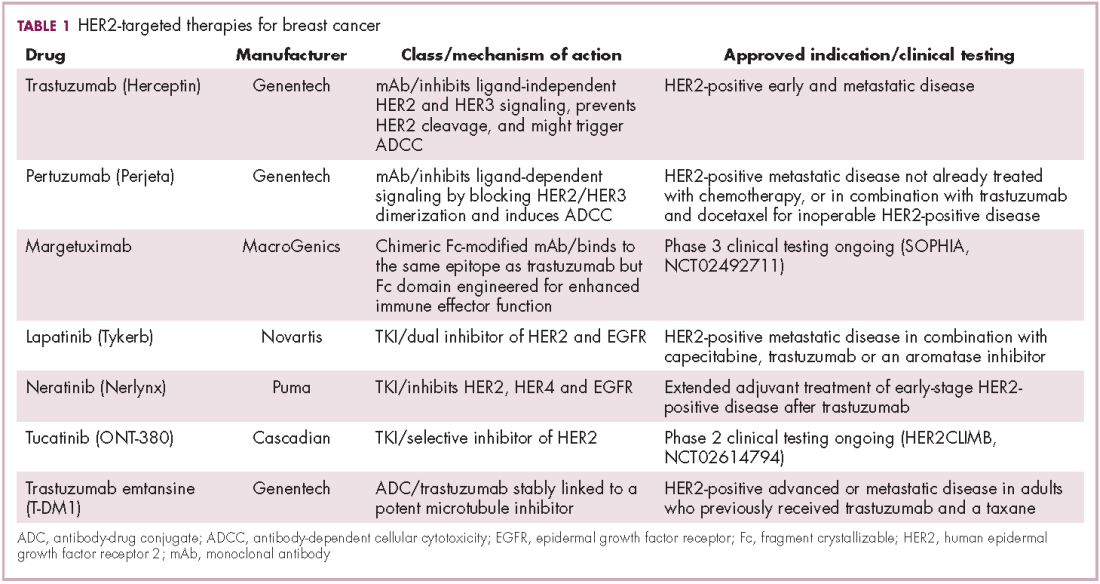
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25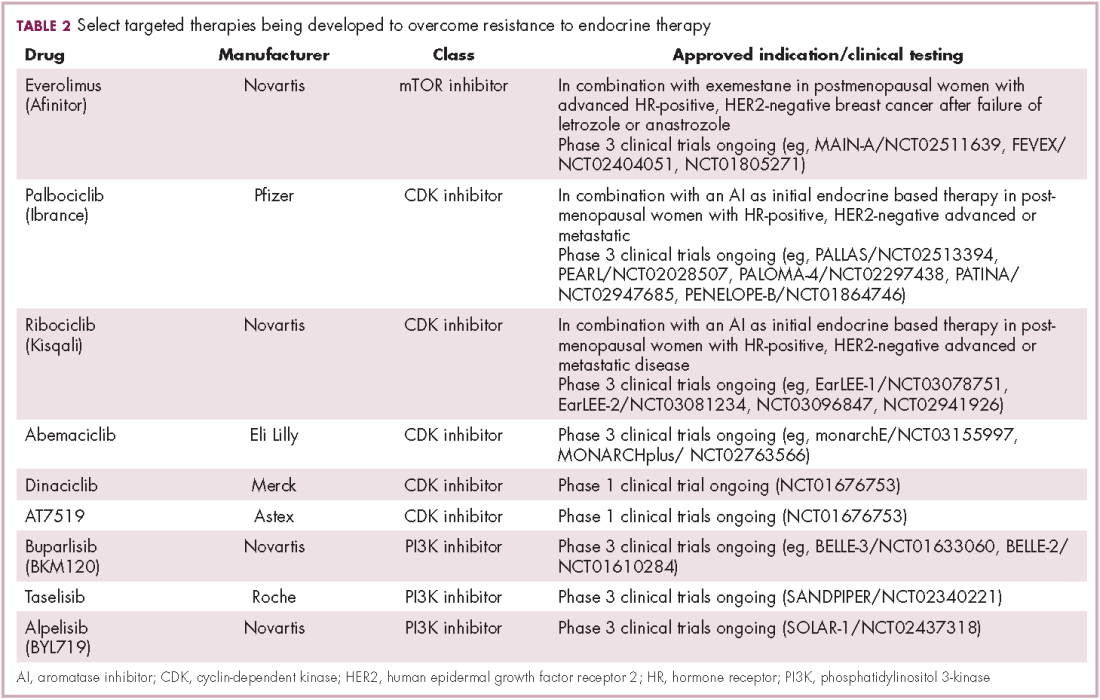
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38