User login
Open Clinical Trials for Patients With Diabetes
Actively Recruiting
→Continuous Glucose Monitoring Devices in Hospitalized Veterans With Diabetes
More than 25% of the patients admitted in the general wards have a history of diabetes mellitus. Up to 30% of the hospitalized diabetics develop hypoglycemia (low glucose values); a condition that is associated with seizures, cardiac arrhythmias, and even death. In veterans, the prevalence is disproportionally higher. It is estimated that 40% to 50% of hospitalized veterans are diabetics. In this clinical trial the investigators describe the development of a novel system, the Glucose Telemetry System, with which glucose values can be wirelessly transmitted from the patient’s bedside to a monitor device at the nursing station. The goal of this work is to develop a more effective glucose surveillance system at the general wards, which can decrease hypoglycemia in the hospital and improve clinical outcomes.
ID: NCT03508934
Sponsor; Investigator: VA Office of Research and Development; Ilias Spanakis, MD
Locations: Baltimore VA Medical Center, Maryland
→Optimizing Gait Rehabilitation for Veterans With Non-Traumatic Lower Limb Amputation (GEM)
The population of older veterans with nontraumatic lower limb amputation is growing. Following lower limb amputation, asymmetrical movements persist during walking and likely contribute to disabling sequelae including secondary pain conditions, poor gait efficiency, impaired physical function, and compromised skin integrity of the residual limb. This study seeks to address chronic gait asymmetry by evaluating the efficacy of two error-manipulation gait training programs to improve gait symmetry for veterans with nontraumatic lower limb amputation. Additionally, this study will evaluate the potential of error-manipulation training programs to improve secondary measures of disability and residual limb skin health. Ultimately, this study aims to improve conventional prosthetic rehabilitation for veterans with nontraumatic amputation through gait training programs based in motor learning principles, resulting in improved.
ID: NCT003995238
Sponsor; Investigator: VA Office of Research and Development; Cory L. Christiansen, PhD
Locations: Rocky Mountain Regional VA Medical Center, Aurora, Colorado; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia
→Empowering Veterans to Actively Communicate and Engage in Shared Decision Making in Medical Visits, A Randomized Controlled Trial (ACTIVet-2)
Type 2 diabetes is a significant condition in VA affecting 20% of VA patients. Adherence to medication regimens and lifestyle factors is important to achieve care goals for these patients. Patients who use active participatory communication behaviors with their providers have better adherence to treatment and better biomedical outcomes, yet many patients are not prepared to engage in active communication with their providers. Existing coaching interventions have not been adopted in practice because of the cost of trained personnel. The investigators haveshown the efficacy of a low-cost video that did not require trained personnel. This proposal proposes to test implementation strategies to deliver that video in VA primary care clinics and to test the effectiveness of the video to improve outcomes in a hybrid type 2 effectiveness implementation trial using a cluster randomized stepped wedgedesign at eight sites. This proposal will test feasibility of implementing the video and if successful will generate the evidence to justify widespread dissemination of the video.
ID: NCT05169359
Sponsor; Investigator: VA Office of Research and Development; Howard S. Gordon, MD
Location: 8 locations, including Jesse Brown VA Medical Center, Chicago; Edward Hines Jr. VA Hopsital, Hines, Illinois
→The Diabetes Staging System in Patient Aligned Care Teams
The purpose of this study is to examine the feasibility/acceptability of the Diabetes Staging System in patient aligned care teams and its ability to increase sodium-glucosecotransporter-2 inhibitor and glucagon-like-1 peptide use in veteran patients with type 2 diabetes and cardiovascular disease and/or chronic kidney disease. A novel type 2 diabetes staging system patterned after Tumor Node Metastasis cancer staging that uses the number of macrovascular and microvascular complications and most recent hemoglobin A1c and glomerular filtration rate to determine Diabetes Staging System stage which reflects disease severity.
ID: NCT06142006
Sponsor; Collaborator: Durham VA Medical Center; Moahad Dar, MD
Location: Greenville VA Health Care Center, North Carolina
→Enhancing Mental and Physical Health of Women Veterans (EMPOWER)
Women veterans are the fastest growing segment of VA users. This dramatic growth has created challenges for VA to ensure that appropriate services are available to meet women veterans’ needs, and that they will want and be able to use those services. The EMPOWER QUERI 2.0 Program is a cluster randomized type 3 hybrid implementation effectiveness trial testing 2 strategies designed to support implementation and sustainment of evidence-based practices for women veterans in at least 20 VA facilities from 4 regions.
ID: NCT05050266
Sponsor; Investigator: VA Office of Research and Development; Alison B. Hamilton, PhD, MPHLocation: VA Greater Los Angeles Healthcare System
→Investigation of Rifampin to Reduce Pedal Amputations for Osteomyelitis in Diabetics (VA INTREPID)
The purpose of this research study is to determine if rifampin, an antibiotic (a medicine that treats infections), is effective in treating osteomyelitis (infection of the bone) of the foot in patients with diabetes. Despite use of powerful antibiotics prescribed over a long period of time, many diabetic patients remain at a high risk for needing an amputation of part of the foot or lower leg because the osteomyelitis is not cured. Some small research studies have shown that addition of rifampin to other antibiotics is effective in treating osteomyelitis. However, because few diabetics with osteomyelitis have been studied, there is no definite proof that it is better than the usual treatments for diabetic patients. If this study finds that adding rifampin to the usual antibiotics prescribed for osteomyelitis reduces the risk for amputations, doctors will be able to more effectively treat many veteran patients with this serious infection. Improving treatment outcomes is an important healthcare goal of the VA.
ID: NCT03012529
Sponsor; Investigator: VA Office of Research and Development; Paul A. Monach, MD, PhD
Location: 30 VA locations, including South Texas Health Care System, San Antonio; Cincinnati VA Medical Center, Ohio; VA Northern California Health Care System, Mather; Washington DC VA Medical Center; William S. Middleton Memorial Veterans Hospital, Madison, Wisconsin
→Effectiveness of Remote Foot Temperature Monitoring (STOP)
Diabetic foot ulcers are common, debilitating, and costly complications of diabetes, disproportionately impacting Black and rural veterans. Forty percent of individuals have an ulcer recurrence within a year of ulcer healing and 65% within 5 years. Monitoring plantar foot temperatures is one of the few interventions that reduces the risk of ulcer recurrence. Despite the evidence, adoption has been poorbecause the original procedures, including the use of handheld thermometers, were burdensome and time-consuming. Podimetrics, a private company, has developed a temperature monitoring system involving a “smart” mat that can wirelessly transmit data and a remote monitoring team that works with VA providers to assist with triage and monitoring. This care model has incredible promise, but has been untested in VA. The investigators propose to conduct a randomized trial to evaluate effectiveness of remote temperature monitoring as well as costs. Additionally, the investigators will evaluate the implementation process, including barriers and facilitators to use among key stakeholders.
ID: NCT05728411
Sponsor; Investigator: VA Office of Research and Development; Rachel M. Thomas
Location: Edward Hines Jr. VA Hospital, Hines, Illinois; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia; VA Puget Sound Health Care System, Seattle, Washington
→Continuous Glucose Monitoring for Hyperglycemia in Critically Ill Patients
The investigators intend to conduct a single-center, prospective, randomized comparative trial of patients admitted to the intensive care unit who received continuous glucose monitoring vs point of care glucose monitoring. The study will examine relevant outcomes for patients in the intensive care unit with diabetes mellitus and/or hyperglycemia. The primary outcome of the study will be the proportion of time in target range (blood glucose 70-180 mg/dL).
ID: NCT05442853
Sponsor; Investigator: Malcom Randall VA
Medical Center; Andrew J. Franck, PharmD
Location: Malcolm Randall VA Medical Center, Gainesville, Florida
→Investigation of Metformin in Pre-Diabetes on Atherosclerotic Cardiovascular OuTcomes (VA-IMPACT)
CSP #2002 is a multicenter, prospective, randomized, double blind, secondary prevention trial to test the hypothesis that treatment with metformin, compared with placebo, reduces mortality and cardiovascular morbidity in veterans with pre-diabetes and established atherosclerotic cardiovascular disease. Qualifying patients have pre-diabetes defined by HbA 1c , fasting blood glucose, or oral glucose tolerance test criteria; clinically evident coronary, cerebrovascular, or peripheral arterial atherosclerotic cardiovascular disease; and estimated glomerular filtration rate of at least 45 mL/min/1.73 m 2 ; and do not fulfill any exclusion criteria.
ID: NCT04838392
Sponsor; Investigator: VA Office of Research and Development; Gregory G. Schwartz, PhD, MD
Locations: 40 locations
Actively Recruiting
→Continuous Glucose Monitoring Devices in Hospitalized Veterans With Diabetes
More than 25% of the patients admitted in the general wards have a history of diabetes mellitus. Up to 30% of the hospitalized diabetics develop hypoglycemia (low glucose values); a condition that is associated with seizures, cardiac arrhythmias, and even death. In veterans, the prevalence is disproportionally higher. It is estimated that 40% to 50% of hospitalized veterans are diabetics. In this clinical trial the investigators describe the development of a novel system, the Glucose Telemetry System, with which glucose values can be wirelessly transmitted from the patient’s bedside to a monitor device at the nursing station. The goal of this work is to develop a more effective glucose surveillance system at the general wards, which can decrease hypoglycemia in the hospital and improve clinical outcomes.
ID: NCT03508934
Sponsor; Investigator: VA Office of Research and Development; Ilias Spanakis, MD
Locations: Baltimore VA Medical Center, Maryland
→Optimizing Gait Rehabilitation for Veterans With Non-Traumatic Lower Limb Amputation (GEM)
The population of older veterans with nontraumatic lower limb amputation is growing. Following lower limb amputation, asymmetrical movements persist during walking and likely contribute to disabling sequelae including secondary pain conditions, poor gait efficiency, impaired physical function, and compromised skin integrity of the residual limb. This study seeks to address chronic gait asymmetry by evaluating the efficacy of two error-manipulation gait training programs to improve gait symmetry for veterans with nontraumatic lower limb amputation. Additionally, this study will evaluate the potential of error-manipulation training programs to improve secondary measures of disability and residual limb skin health. Ultimately, this study aims to improve conventional prosthetic rehabilitation for veterans with nontraumatic amputation through gait training programs based in motor learning principles, resulting in improved.
ID: NCT003995238
Sponsor; Investigator: VA Office of Research and Development; Cory L. Christiansen, PhD
Locations: Rocky Mountain Regional VA Medical Center, Aurora, Colorado; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia
→Empowering Veterans to Actively Communicate and Engage in Shared Decision Making in Medical Visits, A Randomized Controlled Trial (ACTIVet-2)
Type 2 diabetes is a significant condition in VA affecting 20% of VA patients. Adherence to medication regimens and lifestyle factors is important to achieve care goals for these patients. Patients who use active participatory communication behaviors with their providers have better adherence to treatment and better biomedical outcomes, yet many patients are not prepared to engage in active communication with their providers. Existing coaching interventions have not been adopted in practice because of the cost of trained personnel. The investigators haveshown the efficacy of a low-cost video that did not require trained personnel. This proposal proposes to test implementation strategies to deliver that video in VA primary care clinics and to test the effectiveness of the video to improve outcomes in a hybrid type 2 effectiveness implementation trial using a cluster randomized stepped wedgedesign at eight sites. This proposal will test feasibility of implementing the video and if successful will generate the evidence to justify widespread dissemination of the video.
ID: NCT05169359
Sponsor; Investigator: VA Office of Research and Development; Howard S. Gordon, MD
Location: 8 locations, including Jesse Brown VA Medical Center, Chicago; Edward Hines Jr. VA Hopsital, Hines, Illinois
→The Diabetes Staging System in Patient Aligned Care Teams
The purpose of this study is to examine the feasibility/acceptability of the Diabetes Staging System in patient aligned care teams and its ability to increase sodium-glucosecotransporter-2 inhibitor and glucagon-like-1 peptide use in veteran patients with type 2 diabetes and cardiovascular disease and/or chronic kidney disease. A novel type 2 diabetes staging system patterned after Tumor Node Metastasis cancer staging that uses the number of macrovascular and microvascular complications and most recent hemoglobin A1c and glomerular filtration rate to determine Diabetes Staging System stage which reflects disease severity.
ID: NCT06142006
Sponsor; Collaborator: Durham VA Medical Center; Moahad Dar, MD
Location: Greenville VA Health Care Center, North Carolina
→Enhancing Mental and Physical Health of Women Veterans (EMPOWER)
Women veterans are the fastest growing segment of VA users. This dramatic growth has created challenges for VA to ensure that appropriate services are available to meet women veterans’ needs, and that they will want and be able to use those services. The EMPOWER QUERI 2.0 Program is a cluster randomized type 3 hybrid implementation effectiveness trial testing 2 strategies designed to support implementation and sustainment of evidence-based practices for women veterans in at least 20 VA facilities from 4 regions.
ID: NCT05050266
Sponsor; Investigator: VA Office of Research and Development; Alison B. Hamilton, PhD, MPHLocation: VA Greater Los Angeles Healthcare System
→Investigation of Rifampin to Reduce Pedal Amputations for Osteomyelitis in Diabetics (VA INTREPID)
The purpose of this research study is to determine if rifampin, an antibiotic (a medicine that treats infections), is effective in treating osteomyelitis (infection of the bone) of the foot in patients with diabetes. Despite use of powerful antibiotics prescribed over a long period of time, many diabetic patients remain at a high risk for needing an amputation of part of the foot or lower leg because the osteomyelitis is not cured. Some small research studies have shown that addition of rifampin to other antibiotics is effective in treating osteomyelitis. However, because few diabetics with osteomyelitis have been studied, there is no definite proof that it is better than the usual treatments for diabetic patients. If this study finds that adding rifampin to the usual antibiotics prescribed for osteomyelitis reduces the risk for amputations, doctors will be able to more effectively treat many veteran patients with this serious infection. Improving treatment outcomes is an important healthcare goal of the VA.
ID: NCT03012529
Sponsor; Investigator: VA Office of Research and Development; Paul A. Monach, MD, PhD
Location: 30 VA locations, including South Texas Health Care System, San Antonio; Cincinnati VA Medical Center, Ohio; VA Northern California Health Care System, Mather; Washington DC VA Medical Center; William S. Middleton Memorial Veterans Hospital, Madison, Wisconsin
→Effectiveness of Remote Foot Temperature Monitoring (STOP)
Diabetic foot ulcers are common, debilitating, and costly complications of diabetes, disproportionately impacting Black and rural veterans. Forty percent of individuals have an ulcer recurrence within a year of ulcer healing and 65% within 5 years. Monitoring plantar foot temperatures is one of the few interventions that reduces the risk of ulcer recurrence. Despite the evidence, adoption has been poorbecause the original procedures, including the use of handheld thermometers, were burdensome and time-consuming. Podimetrics, a private company, has developed a temperature monitoring system involving a “smart” mat that can wirelessly transmit data and a remote monitoring team that works with VA providers to assist with triage and monitoring. This care model has incredible promise, but has been untested in VA. The investigators propose to conduct a randomized trial to evaluate effectiveness of remote temperature monitoring as well as costs. Additionally, the investigators will evaluate the implementation process, including barriers and facilitators to use among key stakeholders.
ID: NCT05728411
Sponsor; Investigator: VA Office of Research and Development; Rachel M. Thomas
Location: Edward Hines Jr. VA Hospital, Hines, Illinois; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia; VA Puget Sound Health Care System, Seattle, Washington
→Continuous Glucose Monitoring for Hyperglycemia in Critically Ill Patients
The investigators intend to conduct a single-center, prospective, randomized comparative trial of patients admitted to the intensive care unit who received continuous glucose monitoring vs point of care glucose monitoring. The study will examine relevant outcomes for patients in the intensive care unit with diabetes mellitus and/or hyperglycemia. The primary outcome of the study will be the proportion of time in target range (blood glucose 70-180 mg/dL).
ID: NCT05442853
Sponsor; Investigator: Malcom Randall VA
Medical Center; Andrew J. Franck, PharmD
Location: Malcolm Randall VA Medical Center, Gainesville, Florida
→Investigation of Metformin in Pre-Diabetes on Atherosclerotic Cardiovascular OuTcomes (VA-IMPACT)
CSP #2002 is a multicenter, prospective, randomized, double blind, secondary prevention trial to test the hypothesis that treatment with metformin, compared with placebo, reduces mortality and cardiovascular morbidity in veterans with pre-diabetes and established atherosclerotic cardiovascular disease. Qualifying patients have pre-diabetes defined by HbA 1c , fasting blood glucose, or oral glucose tolerance test criteria; clinically evident coronary, cerebrovascular, or peripheral arterial atherosclerotic cardiovascular disease; and estimated glomerular filtration rate of at least 45 mL/min/1.73 m 2 ; and do not fulfill any exclusion criteria.
ID: NCT04838392
Sponsor; Investigator: VA Office of Research and Development; Gregory G. Schwartz, PhD, MD
Locations: 40 locations
Actively Recruiting
→Continuous Glucose Monitoring Devices in Hospitalized Veterans With Diabetes
More than 25% of the patients admitted in the general wards have a history of diabetes mellitus. Up to 30% of the hospitalized diabetics develop hypoglycemia (low glucose values); a condition that is associated with seizures, cardiac arrhythmias, and even death. In veterans, the prevalence is disproportionally higher. It is estimated that 40% to 50% of hospitalized veterans are diabetics. In this clinical trial the investigators describe the development of a novel system, the Glucose Telemetry System, with which glucose values can be wirelessly transmitted from the patient’s bedside to a monitor device at the nursing station. The goal of this work is to develop a more effective glucose surveillance system at the general wards, which can decrease hypoglycemia in the hospital and improve clinical outcomes.
ID: NCT03508934
Sponsor; Investigator: VA Office of Research and Development; Ilias Spanakis, MD
Locations: Baltimore VA Medical Center, Maryland
→Optimizing Gait Rehabilitation for Veterans With Non-Traumatic Lower Limb Amputation (GEM)
The population of older veterans with nontraumatic lower limb amputation is growing. Following lower limb amputation, asymmetrical movements persist during walking and likely contribute to disabling sequelae including secondary pain conditions, poor gait efficiency, impaired physical function, and compromised skin integrity of the residual limb. This study seeks to address chronic gait asymmetry by evaluating the efficacy of two error-manipulation gait training programs to improve gait symmetry for veterans with nontraumatic lower limb amputation. Additionally, this study will evaluate the potential of error-manipulation training programs to improve secondary measures of disability and residual limb skin health. Ultimately, this study aims to improve conventional prosthetic rehabilitation for veterans with nontraumatic amputation through gait training programs based in motor learning principles, resulting in improved.
ID: NCT003995238
Sponsor; Investigator: VA Office of Research and Development; Cory L. Christiansen, PhD
Locations: Rocky Mountain Regional VA Medical Center, Aurora, Colorado; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia
→Empowering Veterans to Actively Communicate and Engage in Shared Decision Making in Medical Visits, A Randomized Controlled Trial (ACTIVet-2)
Type 2 diabetes is a significant condition in VA affecting 20% of VA patients. Adherence to medication regimens and lifestyle factors is important to achieve care goals for these patients. Patients who use active participatory communication behaviors with their providers have better adherence to treatment and better biomedical outcomes, yet many patients are not prepared to engage in active communication with their providers. Existing coaching interventions have not been adopted in practice because of the cost of trained personnel. The investigators haveshown the efficacy of a low-cost video that did not require trained personnel. This proposal proposes to test implementation strategies to deliver that video in VA primary care clinics and to test the effectiveness of the video to improve outcomes in a hybrid type 2 effectiveness implementation trial using a cluster randomized stepped wedgedesign at eight sites. This proposal will test feasibility of implementing the video and if successful will generate the evidence to justify widespread dissemination of the video.
ID: NCT05169359
Sponsor; Investigator: VA Office of Research and Development; Howard S. Gordon, MD
Location: 8 locations, including Jesse Brown VA Medical Center, Chicago; Edward Hines Jr. VA Hopsital, Hines, Illinois
→The Diabetes Staging System in Patient Aligned Care Teams
The purpose of this study is to examine the feasibility/acceptability of the Diabetes Staging System in patient aligned care teams and its ability to increase sodium-glucosecotransporter-2 inhibitor and glucagon-like-1 peptide use in veteran patients with type 2 diabetes and cardiovascular disease and/or chronic kidney disease. A novel type 2 diabetes staging system patterned after Tumor Node Metastasis cancer staging that uses the number of macrovascular and microvascular complications and most recent hemoglobin A1c and glomerular filtration rate to determine Diabetes Staging System stage which reflects disease severity.
ID: NCT06142006
Sponsor; Collaborator: Durham VA Medical Center; Moahad Dar, MD
Location: Greenville VA Health Care Center, North Carolina
→Enhancing Mental and Physical Health of Women Veterans (EMPOWER)
Women veterans are the fastest growing segment of VA users. This dramatic growth has created challenges for VA to ensure that appropriate services are available to meet women veterans’ needs, and that they will want and be able to use those services. The EMPOWER QUERI 2.0 Program is a cluster randomized type 3 hybrid implementation effectiveness trial testing 2 strategies designed to support implementation and sustainment of evidence-based practices for women veterans in at least 20 VA facilities from 4 regions.
ID: NCT05050266
Sponsor; Investigator: VA Office of Research and Development; Alison B. Hamilton, PhD, MPHLocation: VA Greater Los Angeles Healthcare System
→Investigation of Rifampin to Reduce Pedal Amputations for Osteomyelitis in Diabetics (VA INTREPID)
The purpose of this research study is to determine if rifampin, an antibiotic (a medicine that treats infections), is effective in treating osteomyelitis (infection of the bone) of the foot in patients with diabetes. Despite use of powerful antibiotics prescribed over a long period of time, many diabetic patients remain at a high risk for needing an amputation of part of the foot or lower leg because the osteomyelitis is not cured. Some small research studies have shown that addition of rifampin to other antibiotics is effective in treating osteomyelitis. However, because few diabetics with osteomyelitis have been studied, there is no definite proof that it is better than the usual treatments for diabetic patients. If this study finds that adding rifampin to the usual antibiotics prescribed for osteomyelitis reduces the risk for amputations, doctors will be able to more effectively treat many veteran patients with this serious infection. Improving treatment outcomes is an important healthcare goal of the VA.
ID: NCT03012529
Sponsor; Investigator: VA Office of Research and Development; Paul A. Monach, MD, PhD
Location: 30 VA locations, including South Texas Health Care System, San Antonio; Cincinnati VA Medical Center, Ohio; VA Northern California Health Care System, Mather; Washington DC VA Medical Center; William S. Middleton Memorial Veterans Hospital, Madison, Wisconsin
→Effectiveness of Remote Foot Temperature Monitoring (STOP)
Diabetic foot ulcers are common, debilitating, and costly complications of diabetes, disproportionately impacting Black and rural veterans. Forty percent of individuals have an ulcer recurrence within a year of ulcer healing and 65% within 5 years. Monitoring plantar foot temperatures is one of the few interventions that reduces the risk of ulcer recurrence. Despite the evidence, adoption has been poorbecause the original procedures, including the use of handheld thermometers, were burdensome and time-consuming. Podimetrics, a private company, has developed a temperature monitoring system involving a “smart” mat that can wirelessly transmit data and a remote monitoring team that works with VA providers to assist with triage and monitoring. This care model has incredible promise, but has been untested in VA. The investigators propose to conduct a randomized trial to evaluate effectiveness of remote temperature monitoring as well as costs. Additionally, the investigators will evaluate the implementation process, including barriers and facilitators to use among key stakeholders.
ID: NCT05728411
Sponsor; Investigator: VA Office of Research and Development; Rachel M. Thomas
Location: Edward Hines Jr. VA Hospital, Hines, Illinois; Hunter Holmes McGuire VA Medical Center, Richmond, Virginia; VA Puget Sound Health Care System, Seattle, Washington
→Continuous Glucose Monitoring for Hyperglycemia in Critically Ill Patients
The investigators intend to conduct a single-center, prospective, randomized comparative trial of patients admitted to the intensive care unit who received continuous glucose monitoring vs point of care glucose monitoring. The study will examine relevant outcomes for patients in the intensive care unit with diabetes mellitus and/or hyperglycemia. The primary outcome of the study will be the proportion of time in target range (blood glucose 70-180 mg/dL).
ID: NCT05442853
Sponsor; Investigator: Malcom Randall VA
Medical Center; Andrew J. Franck, PharmD
Location: Malcolm Randall VA Medical Center, Gainesville, Florida
→Investigation of Metformin in Pre-Diabetes on Atherosclerotic Cardiovascular OuTcomes (VA-IMPACT)
CSP #2002 is a multicenter, prospective, randomized, double blind, secondary prevention trial to test the hypothesis that treatment with metformin, compared with placebo, reduces mortality and cardiovascular morbidity in veterans with pre-diabetes and established atherosclerotic cardiovascular disease. Qualifying patients have pre-diabetes defined by HbA 1c , fasting blood glucose, or oral glucose tolerance test criteria; clinically evident coronary, cerebrovascular, or peripheral arterial atherosclerotic cardiovascular disease; and estimated glomerular filtration rate of at least 45 mL/min/1.73 m 2 ; and do not fulfill any exclusion criteria.
ID: NCT04838392
Sponsor; Investigator: VA Office of Research and Development; Gregory G. Schwartz, PhD, MD
Locations: 40 locations
Open Clinical Trials for Patients With Prostate Cancer
The clinical trials listed below are all open as of July 12, 2024; have ≥ 1 US Department of Veterans Affairs (VA) medical center (VAMC) or US Department of Defense (DoD) military treatment facility location recruiting patients; and are focused on treatments for prostate cancer. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Actively Recruiting
Patient Decision Making About Precision Oncology in Veterans With Advanced Prostate Cancer
This clinical trial explores and implements methods to improve informed decision making regarding precision oncology tests among veterans with prostate cancer that may have spread from where it first started to nearby tissue, lymph nodes, or distant parts of the body (advanced). Precision oncology, the use of germline genetic testing and tumor-based molecular assays to inform cancer care, has become an important aspect of evidence-based care for men with advanced prostate cancer. Veterans with metastatic castrate-resistant prostate cancer may not be carrying out informed decision making due to unmet decisional needs. An informed decision is a choice based on complete and accurate information. The information gained from this study will help researchers develop a decision support intervention and implement the intervention. A decision support intervention may serve as a valuable tool to reduce ongoing racial disparities in genetic testing and encourage enrollment to precision oncology trials.
ID: NCT05396872
Sponsor; Collaborator: University of California, San Francisco; DoD
Location: San Francisco VAMC
DeADT - Living Well With Prostate Cancer
The goal of this pilot randomized implementation trial is to compare 2 strategies to reduce low-value androgen deprivation therapy (ADT) use for prostate cancer care. The aim of the study is to compare implementation of the 2 strategies: use of a clinical reminder order check intervention vs a clinician script/patient education approach, and their impacts on low-value ADT use after 6 months. The main goal of both interventions will be to decrease ADT overuse for patients with prostate cancer, but to do this in a way that is acceptable to the provider who treat these patients. Provider participants will engage with 1 of the interventions triggered in the electronic health record when their patients are deemed likely to receive low-value ADT. Provider participants receive only 1 intervention. The intervention is triggered for every clinic visit involving a patient deemed to be receiving low-value ADT, so provider participants may receive their assigned intervention multiple times. Researchers will compare provider use of both strategies to determine implementation outcomes and whether 1 was more effective in reducing low-value ADT use.
ID: NCT06199986
Sponsor; Collaborator: University of Michigan; VA, National Cancer Institute
Location: VA Ann Arbor Healthcare System
VA Seamless Phase II/III Randomized Trial of Standard Systemic Therapy With or Without PET-Directed Local Therapy for Oligometastatic Prostate Cancer (VA STARPORT)
This is a prospective, open-label, multicenter, seamless phase II to phase III randomized clinical trial designed to compare somatostatin with or without positron emission tomography (PET)-directed local therapy in improving the castration-resistant prostate cancer-free survival for veterans with oligometastatic prostate cancer. Oligometastasis will be defined as 1 to 10 sites of metastatic disease based on the clinical determination.
ID: NCT04787744
Sponsor; Investigators: VA Office of Research and Development; Abhishek Solani, MD, MS, Edward Hines Jr.
Locations: VA Long Beach Healthcare System, VA Greater Los Angeles Healthcare System, Bay Pines VA Healthcare System, Edward Hines Jr. VA Hospital, Richard L. Roudebush VAMC, Baltimore VAMC, VA Boston Healthcare System, VA Ann Arbor Healthcare System, Minneapolis VA Health Care System, Kansas City VAMC, VA New Jersey Healthcare System, VA NY Harbor Healthcare System, Durham VAMC, Louis Stokes VAMC, Corporal Michael J. Crescenz VAMC, Michael E. DeBakey VAMC, Hunter Holmes McGuire VAMC, William S. Middleton Memorial Veterans Hospital, Clement J. Zablocki VAMC
The Prostate Cancer, Genetic Risk, and Equitable Screening Study (ProGRESS)
Prostate cancer is the most common non-skin cancer among veterans and the second leading cause of male cancer death. Current methods of screening men for prostate cancer are inaccurate and cannot identify which men do not have prostate cancer or have low-grade cases that will not cause harm and which men have significant prostate cancer needing treatment. False-positive screening tests can result in unnecessary prostate biopsies for men who do not need them. However, new genetic testing might help identify which men are at highest risk for prostate cancer. This study will examine whether a genetic test helps identify men at risk for significant prostate cancer while helping men who are at low risk for prostate cancer avoid unnecessary biopsies. If this genetic test proves beneficial, it will improve the way that health care providers screen male veterans for prostate cancer.
ID: NCT05926102
Sponsor; Investigator: VA Office of Research and Development; Jason L. Vassy, MD, MPH
Location: VA Boston Healthcare System
Prostate Active Surveillance Study (PASS)
This research study is for men who have chosen active surveillance as a management plan for their prostate cancer. Active surveillance is defined as close monitoring of prostate cancer with the offer of treatment if there are changes in test results. This study seeks to discover markers that will identify cancers that are more aggressive from those tumors that grow slowly.
ID: NCT00756665
Sponsor; Collaborators: University of Washington; Canary Foundation, Early Detection Research Network
Locations: VA San Francisco Health Care System, VA Puget Sound Health Care System
A Study of Checkpoint Inhibitors in Men With Progressive Metastatic Castrate Resistant Prostate Cancer Characterized by a Mismatch Repair Deficiency or Biallelic CDK12 Inactivation (CHOMP)
The primary objective is to assess the activity and efficacy of pembrolizumab, a checkpoint inhibitor, in veterans with metastatic castration-resistant prostate cancer characterized by either mismatch repair deficiency (dMMR) or biallelic inactivation of CDK12 (CDK12-/-). The secondary objectives involve determining the frequency with which dMMR and CDK12-/- occur in this patient population, as well as the effects of pembrolizumab on various clinical endpoints (time to prostate-specific antigen progression, maximal prostate-specific antigen response, time to initiation of alternative antineoplastic therapy, time to radiographic progression, overall survival, and safety and tolerability). Lastly, the study will compare the pretreatment and at-progression metastatic tumor biopsies to investigate the molecular correlates of resistance and sensitivity to pembrolizumab via RNA-sequencing, exome-sequencing, selected protein analyses, and multiplexed immunofluorescence.
ID: NCT04104893
Sponsor; Collaborator: VA Office of Research and Development; Merck Sharp & Dohme LLC
Locations: San Francisco VAMC, VA Greater Los Angeles Healthcare System, Washington DC VAMC, Bay Pines VA Healthcare System Jesse Brown VAMC, VA Ann Arbor Healthcare System, James J. Peter VAMC, VA NY Harbor Healthcare System, Durham VAMC, Corporal Michael J. Crescenz VAMC, Hunter Holmes McGuire VAMC, VA Puget Sound Health Care System
A Single-Arm Phase II Study of Neoadjuvant Intensified Androgen Deprivation (Leuprolideand Abiraterone Acetate) in Combination With AKT Inhibition (Capivasertib) for High-Risk Localized Prostate Cancer With PTEN Loss (SNARE)
The purpose of this study is to learn about how an investigational drug intervention completed before doing prostate surgery (specifically, radical prostatectomy with lymph node dissection) may help in the treatment of high-risk localized prostate cancers that are most resistant to standard treatments. This is a phase II research study. For this study, capivasertib, the study drug, will be taken with intensified androgen deprivation therapy drugs (iADT; abiraterone and leuprolide) prior to radical prostatectomy. This study drug treatment will be evaluated to see if it is effective in shrinking and destroying prostate cancer tumors prior to surgery and to further evaluate its safety prior to prostate cancer surgery.
ID: NCT05593497
Sponsor; Investigator: VA Office of Research and Development; Ryan P. Kopp, MD
Locations: VA Greater Los Angeles Healthcare System, James J. Peters VAMC, VA Portland Health Care System, South Texas Veterans Health Care System, VA Puget Sound Health Care System
Active, Not Recruiting
Intramuscular Mechanisms of Androgen Deprivation-related Sarcopenia
Prostate cancer is the most common cancer among men and is even more common in the military and veteran population. For patients with advanced prostate cancer, the most common treatment includes androgen deprivation therapy (ADT), or the lowering of the levels of the hormone testosterone as much as possible. Unfortunately, ADT also causes patients to be fatigued, weak, and to lose muscle. This is often referred to as “sarcopenia,” and it leads to falls, poor quality of life, and higher risk of death. Currently, there is no treatment for sarcopenia because the investigators do not understand the mechanisms that cause it. The mitochondria are part of the cells responsible for providing energy to muscles, but to this date, the investigators do not know if it is affected in prostate cancer patients with sarcopenia due to ADT.
ID: NCT03867357
Sponsor; Collaborators: Seattle Institute for Biomedical and Clinical Research; DoD, University of Washington
Location: VA Puget Sound Health Care System
Radiation Therapy With or Without Androgen-Deprivation Therapy in Treating Patients With Prostate Cancer
RATIONALE: Radiation therapy uses high-energy X-rays and other types of radiation to kill tumor cells and shrink tumors. Androgens can cause the growth of prostate cancer cells. Androgen deprivation therapy (ADT) may lessen the amount of androgens made by the body. It is not yet known whether radiation therapy is more effective with or without ADT in treating patients with prostate cancer.
PURPOSE: This randomized phase III trial is studying radiation therapy to see how well it works compared with radiation therapy given together with ADT in treating patients with prostate cancer.
ID: NCT00936390
Sponsor; Collaborators: Radiation Therapy Oncology Group; National Cancer Institute, NRG Oncology
Locations: 518 locations, James A. Haley VA Hospital
Enzalutamide With or Without Abiraterone and Prednisone in Treating Patients With Castration-Resistant Metastatic Prostate Cancer
This randomized phase III trial studies enzalutamide to see how well it works compared to enzalutamide, abiraterone, and prednisone in treating patients with castration-resistant metastatic prostate cancer. Androgens can cause the growth of prostate cancer cells. Drugs, such as enzalutamide, abiraterone acetate, and prednisone, may lessen the amount of androgens made by the body.
ID: NCT01949337
Sponsor; Collaborators: Alliance for Clinical Trials in Oncology; NCI, Astellas Pharma US, Inc., Medivation, Inc., Biologics, Inc.
Locations: 539 locations, including VA Connecticut Healthcare System
S1216, Phase III ADT+TAK-700 vs ADT+Bicalutamide for Metastatic Prostate Cancer (S1216)
The purpose of this study is to compare overall survival in newly diagnosed metastatic prostate cancer patients randomly assigned to ADT + TAK-700 vs androgen deprivation therapy (ADT) + bicalutamide.
ID: NCT01809691
Sponsor; Collaborators: SWOG Cancer Research Network; Millennium Pharmaceuticals, Inc., NCI
Locations: 560 locations, including VA New York Harbor Healthcare System
Androgen Ablation Therapy With or Without Chemotherapy in Treating Patients With Metastatic Prostate Cancer (CHAARTED)
RATIONALE: Androgens can cause the growth of prostate cancer cells. Androgen ablation therapy may stop the adrenal glands from making androgens. Drugs used in chemotherapy, such as docetaxel, work in different ways to stop the growth of tumor cells, either by killing the cells or by stopping them from dividing. It is not yet known whether androgen-ablation therapy is more effective with or without docetaxel in treating metastatic prostate cancer.
PURPOSE: This randomized phase III trial is studying androgen ablation therapy and chemotherapy to see how well they work compared to androgen ablation therapy alone in treating patients with metastatic prostate cancer.
ID: NCT00309985
Sponsor; Collaborator: ECOG-ACRIN Cancer Research Group; NCI
Locations: 343 locations, including Mather VAMC
Not Yet Recruiting
biraterone, Enzalutamide, or Apalutamide in Castrate-Sensitive Prostate Cancer
The investigators have used national Veterans Health Administration (VHA) data to demonstrate real-world efficacy of abiraterone and enzalutamide in veterans with metastatic castration-resistant prostate cancer. In the real world that is the VHA, the investigators have successfully estimated g values that accurately predict overall survival, and the use of this metric in other settings should now be explored. In the egalitarian system that is the VHA, the treatment of prostate cancer is excellent, uniform across the US and indifferent to race. The choices made are clearly personalized, given not all men received all therapies, and younger veterans were treated more aggressively.
ID: NCT05422911
Sponsor: James J. Peters VAMC
Location: James J. Peters VAMC
18F-DCFPyL PET/CT Impact on Treatment Strategies for Patients With Prostate Cancer (PROSPYL)
The main purpose of this phase II trial study is to determine whether a positron emission tomography (PET)/computed tomography (CT) scan using 18F-DCFPyL affects the clinical management plan in veterans. In this study, the management plan prior to and after 18F-DCFPyL PET/CT will be recorded by specific questionnaires, and corresponding changes in management will be analyzed. The scan will be used to see how the disease has spread. Both the treatment strategies and probable disease outcomes as relevant to clinical endpoints will be assessed. This study is open to veterans only.
ID: NCT04390880
Sponsor, Investigator: VA Greater Los Angeles Healthcare System; Gholam Berenji, MD
Location: VA Greater Los Angeles Healthcare System
18F-DCFPyL PET-CT Scan and Prostate Cancer
The primary objective of this study is to assess the efficacy of 18F-DCFPyL PET-CT for initial staging of prostate cancer in veterans compared to conventional imaging (99mTc-MDP bone scan and diagnostic CT or MRI). The primary clinical endpoint of our study is the percentage of veterans with prostate cancer in which the 18F-DCFPyL PET-CT identifies M1 disease at initial staging. Secondary objectives included frequency of the change in primary treatment plan after initial staging.
ID: NCT03852654
Sponsor, Investigator: Lida Jafari, MD
Location: VA Greater Los Angeles Healthcare System
Neoadjuvant Therapy With Docetaxel and Ketoconazole in Patients With High-Risk Prostate Cancer: A Pilot Study (IST 16167)
Eligible patients with high-risk prostate cancer who are scheduled to undergo radical prostatectomy will receive 4 cycles of therapy with ketoconazole and docetaxel prior to surgery resection. A cycle of therapy is defined as 21 days (3 weeks). Pharmacokinetic analysis will be performed during the first and second cycles of therapy. All patients will be evaluated for toxicity, tumor response, and recurrence.
ID: NCT00870714
Sponsor, Collaborator: Kansas City VAMC; Sanofi
Location: Kansas City VAMC
A Study of Epirubicin With Estramustine Phosphate and Celecoxib for the Treatment of Prostate Cancer
The purpose of this clinical trial is to find out the effect of epirubicin with estramustine phosphate and celecoxib on PSA and objective response in patients with hormone-resistant prostate cancer, as well as to evaluate the toxicity and quality of life of this combination. Celecoxib is an FDA-approved drug that treats arthritis. Epirubicin, alone or with estramustine phosphate, has been used in the treatment of hormone-resistant prostate cancer. These drugs have demonstrated evidence of tumor blood vessel suppression and a combination of these 3 drugs could possibly arrest further tumor growth or even make the tumor decrease in size.
ID: NCT00218205
Sponsor, Collaborator; Investigator: VA New Jersey Health Care System; Pfizer; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Phase II Trial of Combination Therapy With Celecoxib and Taxotere for the Treatment of Stage D3 Prostate Cancer
The purpose of this clinical trial is to find out the safety and effectiveness as well as the patient’s quality of life while taking the combination of Taxotere and celecoxib on patients with hormone refractory prostate cancer. Celecoxib (Celebrex) is an FDA-approved drug that treats arthritis. Taxotere (Docetaxel) is an FDA-approved chemotherapy drug to treat certain forms of cancer. Both drugs have demonstrated evidences of tumor blood vessel suppression and combination of these 2 drugs could possibly arrest further tumor growth or make the tumor decrease in size.
ID: NCT00215345
Sponsor, Collaborator; Investigator: Department of Veterans Affairs, New Jersey; Pfizer, Sanofi; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Yoga Program for Patients Undergoing Prostate Cancer Surgery
Men with localized prostate cancer are often treated with surgery, a treatment that is associated with high rates of adverse effects such as erectile dysfunction (ED) and urinary incontinence (UI) which impact quality of life. Yoga may improve control of UI and improve ED by bringing awareness to and strengthening the pelvic floor musculature. The randomized controlled pilot study is to assess the feasibility of an innovative hybrid (in-person and virtual) twice-weekly yoga program that includes a prehabilitation component and to obtain preliminary data that will help assess its potential effectiveness in alleviating prostate cancer treatment symptom burden (primarily ED and UI). The long-term goal is to develop a scalable and sustainable yoga program that helps cancer survivors manage their treatment side effects.
ID: NCT05929300
Sponsor, Investigator: VA Office of Research and Development; Abigail Silva, PhD, MPH
Location: Edward Hines Jr. VA Hospital
The clinical trials listed below are all open as of July 12, 2024; have ≥ 1 US Department of Veterans Affairs (VA) medical center (VAMC) or US Department of Defense (DoD) military treatment facility location recruiting patients; and are focused on treatments for prostate cancer. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Actively Recruiting
Patient Decision Making About Precision Oncology in Veterans With Advanced Prostate Cancer
This clinical trial explores and implements methods to improve informed decision making regarding precision oncology tests among veterans with prostate cancer that may have spread from where it first started to nearby tissue, lymph nodes, or distant parts of the body (advanced). Precision oncology, the use of germline genetic testing and tumor-based molecular assays to inform cancer care, has become an important aspect of evidence-based care for men with advanced prostate cancer. Veterans with metastatic castrate-resistant prostate cancer may not be carrying out informed decision making due to unmet decisional needs. An informed decision is a choice based on complete and accurate information. The information gained from this study will help researchers develop a decision support intervention and implement the intervention. A decision support intervention may serve as a valuable tool to reduce ongoing racial disparities in genetic testing and encourage enrollment to precision oncology trials.
ID: NCT05396872
Sponsor; Collaborator: University of California, San Francisco; DoD
Location: San Francisco VAMC
DeADT - Living Well With Prostate Cancer
The goal of this pilot randomized implementation trial is to compare 2 strategies to reduce low-value androgen deprivation therapy (ADT) use for prostate cancer care. The aim of the study is to compare implementation of the 2 strategies: use of a clinical reminder order check intervention vs a clinician script/patient education approach, and their impacts on low-value ADT use after 6 months. The main goal of both interventions will be to decrease ADT overuse for patients with prostate cancer, but to do this in a way that is acceptable to the provider who treat these patients. Provider participants will engage with 1 of the interventions triggered in the electronic health record when their patients are deemed likely to receive low-value ADT. Provider participants receive only 1 intervention. The intervention is triggered for every clinic visit involving a patient deemed to be receiving low-value ADT, so provider participants may receive their assigned intervention multiple times. Researchers will compare provider use of both strategies to determine implementation outcomes and whether 1 was more effective in reducing low-value ADT use.
ID: NCT06199986
Sponsor; Collaborator: University of Michigan; VA, National Cancer Institute
Location: VA Ann Arbor Healthcare System
VA Seamless Phase II/III Randomized Trial of Standard Systemic Therapy With or Without PET-Directed Local Therapy for Oligometastatic Prostate Cancer (VA STARPORT)
This is a prospective, open-label, multicenter, seamless phase II to phase III randomized clinical trial designed to compare somatostatin with or without positron emission tomography (PET)-directed local therapy in improving the castration-resistant prostate cancer-free survival for veterans with oligometastatic prostate cancer. Oligometastasis will be defined as 1 to 10 sites of metastatic disease based on the clinical determination.
ID: NCT04787744
Sponsor; Investigators: VA Office of Research and Development; Abhishek Solani, MD, MS, Edward Hines Jr.
Locations: VA Long Beach Healthcare System, VA Greater Los Angeles Healthcare System, Bay Pines VA Healthcare System, Edward Hines Jr. VA Hospital, Richard L. Roudebush VAMC, Baltimore VAMC, VA Boston Healthcare System, VA Ann Arbor Healthcare System, Minneapolis VA Health Care System, Kansas City VAMC, VA New Jersey Healthcare System, VA NY Harbor Healthcare System, Durham VAMC, Louis Stokes VAMC, Corporal Michael J. Crescenz VAMC, Michael E. DeBakey VAMC, Hunter Holmes McGuire VAMC, William S. Middleton Memorial Veterans Hospital, Clement J. Zablocki VAMC
The Prostate Cancer, Genetic Risk, and Equitable Screening Study (ProGRESS)
Prostate cancer is the most common non-skin cancer among veterans and the second leading cause of male cancer death. Current methods of screening men for prostate cancer are inaccurate and cannot identify which men do not have prostate cancer or have low-grade cases that will not cause harm and which men have significant prostate cancer needing treatment. False-positive screening tests can result in unnecessary prostate biopsies for men who do not need them. However, new genetic testing might help identify which men are at highest risk for prostate cancer. This study will examine whether a genetic test helps identify men at risk for significant prostate cancer while helping men who are at low risk for prostate cancer avoid unnecessary biopsies. If this genetic test proves beneficial, it will improve the way that health care providers screen male veterans for prostate cancer.
ID: NCT05926102
Sponsor; Investigator: VA Office of Research and Development; Jason L. Vassy, MD, MPH
Location: VA Boston Healthcare System
Prostate Active Surveillance Study (PASS)
This research study is for men who have chosen active surveillance as a management plan for their prostate cancer. Active surveillance is defined as close monitoring of prostate cancer with the offer of treatment if there are changes in test results. This study seeks to discover markers that will identify cancers that are more aggressive from those tumors that grow slowly.
ID: NCT00756665
Sponsor; Collaborators: University of Washington; Canary Foundation, Early Detection Research Network
Locations: VA San Francisco Health Care System, VA Puget Sound Health Care System
A Study of Checkpoint Inhibitors in Men With Progressive Metastatic Castrate Resistant Prostate Cancer Characterized by a Mismatch Repair Deficiency or Biallelic CDK12 Inactivation (CHOMP)
The primary objective is to assess the activity and efficacy of pembrolizumab, a checkpoint inhibitor, in veterans with metastatic castration-resistant prostate cancer characterized by either mismatch repair deficiency (dMMR) or biallelic inactivation of CDK12 (CDK12-/-). The secondary objectives involve determining the frequency with which dMMR and CDK12-/- occur in this patient population, as well as the effects of pembrolizumab on various clinical endpoints (time to prostate-specific antigen progression, maximal prostate-specific antigen response, time to initiation of alternative antineoplastic therapy, time to radiographic progression, overall survival, and safety and tolerability). Lastly, the study will compare the pretreatment and at-progression metastatic tumor biopsies to investigate the molecular correlates of resistance and sensitivity to pembrolizumab via RNA-sequencing, exome-sequencing, selected protein analyses, and multiplexed immunofluorescence.
ID: NCT04104893
Sponsor; Collaborator: VA Office of Research and Development; Merck Sharp & Dohme LLC
Locations: San Francisco VAMC, VA Greater Los Angeles Healthcare System, Washington DC VAMC, Bay Pines VA Healthcare System Jesse Brown VAMC, VA Ann Arbor Healthcare System, James J. Peter VAMC, VA NY Harbor Healthcare System, Durham VAMC, Corporal Michael J. Crescenz VAMC, Hunter Holmes McGuire VAMC, VA Puget Sound Health Care System
A Single-Arm Phase II Study of Neoadjuvant Intensified Androgen Deprivation (Leuprolideand Abiraterone Acetate) in Combination With AKT Inhibition (Capivasertib) for High-Risk Localized Prostate Cancer With PTEN Loss (SNARE)
The purpose of this study is to learn about how an investigational drug intervention completed before doing prostate surgery (specifically, radical prostatectomy with lymph node dissection) may help in the treatment of high-risk localized prostate cancers that are most resistant to standard treatments. This is a phase II research study. For this study, capivasertib, the study drug, will be taken with intensified androgen deprivation therapy drugs (iADT; abiraterone and leuprolide) prior to radical prostatectomy. This study drug treatment will be evaluated to see if it is effective in shrinking and destroying prostate cancer tumors prior to surgery and to further evaluate its safety prior to prostate cancer surgery.
ID: NCT05593497
Sponsor; Investigator: VA Office of Research and Development; Ryan P. Kopp, MD
Locations: VA Greater Los Angeles Healthcare System, James J. Peters VAMC, VA Portland Health Care System, South Texas Veterans Health Care System, VA Puget Sound Health Care System
Active, Not Recruiting
Intramuscular Mechanisms of Androgen Deprivation-related Sarcopenia
Prostate cancer is the most common cancer among men and is even more common in the military and veteran population. For patients with advanced prostate cancer, the most common treatment includes androgen deprivation therapy (ADT), or the lowering of the levels of the hormone testosterone as much as possible. Unfortunately, ADT also causes patients to be fatigued, weak, and to lose muscle. This is often referred to as “sarcopenia,” and it leads to falls, poor quality of life, and higher risk of death. Currently, there is no treatment for sarcopenia because the investigators do not understand the mechanisms that cause it. The mitochondria are part of the cells responsible for providing energy to muscles, but to this date, the investigators do not know if it is affected in prostate cancer patients with sarcopenia due to ADT.
ID: NCT03867357
Sponsor; Collaborators: Seattle Institute for Biomedical and Clinical Research; DoD, University of Washington
Location: VA Puget Sound Health Care System
Radiation Therapy With or Without Androgen-Deprivation Therapy in Treating Patients With Prostate Cancer
RATIONALE: Radiation therapy uses high-energy X-rays and other types of radiation to kill tumor cells and shrink tumors. Androgens can cause the growth of prostate cancer cells. Androgen deprivation therapy (ADT) may lessen the amount of androgens made by the body. It is not yet known whether radiation therapy is more effective with or without ADT in treating patients with prostate cancer.
PURPOSE: This randomized phase III trial is studying radiation therapy to see how well it works compared with radiation therapy given together with ADT in treating patients with prostate cancer.
ID: NCT00936390
Sponsor; Collaborators: Radiation Therapy Oncology Group; National Cancer Institute, NRG Oncology
Locations: 518 locations, James A. Haley VA Hospital
Enzalutamide With or Without Abiraterone and Prednisone in Treating Patients With Castration-Resistant Metastatic Prostate Cancer
This randomized phase III trial studies enzalutamide to see how well it works compared to enzalutamide, abiraterone, and prednisone in treating patients with castration-resistant metastatic prostate cancer. Androgens can cause the growth of prostate cancer cells. Drugs, such as enzalutamide, abiraterone acetate, and prednisone, may lessen the amount of androgens made by the body.
ID: NCT01949337
Sponsor; Collaborators: Alliance for Clinical Trials in Oncology; NCI, Astellas Pharma US, Inc., Medivation, Inc., Biologics, Inc.
Locations: 539 locations, including VA Connecticut Healthcare System
S1216, Phase III ADT+TAK-700 vs ADT+Bicalutamide for Metastatic Prostate Cancer (S1216)
The purpose of this study is to compare overall survival in newly diagnosed metastatic prostate cancer patients randomly assigned to ADT + TAK-700 vs androgen deprivation therapy (ADT) + bicalutamide.
ID: NCT01809691
Sponsor; Collaborators: SWOG Cancer Research Network; Millennium Pharmaceuticals, Inc., NCI
Locations: 560 locations, including VA New York Harbor Healthcare System
Androgen Ablation Therapy With or Without Chemotherapy in Treating Patients With Metastatic Prostate Cancer (CHAARTED)
RATIONALE: Androgens can cause the growth of prostate cancer cells. Androgen ablation therapy may stop the adrenal glands from making androgens. Drugs used in chemotherapy, such as docetaxel, work in different ways to stop the growth of tumor cells, either by killing the cells or by stopping them from dividing. It is not yet known whether androgen-ablation therapy is more effective with or without docetaxel in treating metastatic prostate cancer.
PURPOSE: This randomized phase III trial is studying androgen ablation therapy and chemotherapy to see how well they work compared to androgen ablation therapy alone in treating patients with metastatic prostate cancer.
ID: NCT00309985
Sponsor; Collaborator: ECOG-ACRIN Cancer Research Group; NCI
Locations: 343 locations, including Mather VAMC
Not Yet Recruiting
biraterone, Enzalutamide, or Apalutamide in Castrate-Sensitive Prostate Cancer
The investigators have used national Veterans Health Administration (VHA) data to demonstrate real-world efficacy of abiraterone and enzalutamide in veterans with metastatic castration-resistant prostate cancer. In the real world that is the VHA, the investigators have successfully estimated g values that accurately predict overall survival, and the use of this metric in other settings should now be explored. In the egalitarian system that is the VHA, the treatment of prostate cancer is excellent, uniform across the US and indifferent to race. The choices made are clearly personalized, given not all men received all therapies, and younger veterans were treated more aggressively.
ID: NCT05422911
Sponsor: James J. Peters VAMC
Location: James J. Peters VAMC
18F-DCFPyL PET/CT Impact on Treatment Strategies for Patients With Prostate Cancer (PROSPYL)
The main purpose of this phase II trial study is to determine whether a positron emission tomography (PET)/computed tomography (CT) scan using 18F-DCFPyL affects the clinical management plan in veterans. In this study, the management plan prior to and after 18F-DCFPyL PET/CT will be recorded by specific questionnaires, and corresponding changes in management will be analyzed. The scan will be used to see how the disease has spread. Both the treatment strategies and probable disease outcomes as relevant to clinical endpoints will be assessed. This study is open to veterans only.
ID: NCT04390880
Sponsor, Investigator: VA Greater Los Angeles Healthcare System; Gholam Berenji, MD
Location: VA Greater Los Angeles Healthcare System
18F-DCFPyL PET-CT Scan and Prostate Cancer
The primary objective of this study is to assess the efficacy of 18F-DCFPyL PET-CT for initial staging of prostate cancer in veterans compared to conventional imaging (99mTc-MDP bone scan and diagnostic CT or MRI). The primary clinical endpoint of our study is the percentage of veterans with prostate cancer in which the 18F-DCFPyL PET-CT identifies M1 disease at initial staging. Secondary objectives included frequency of the change in primary treatment plan after initial staging.
ID: NCT03852654
Sponsor, Investigator: Lida Jafari, MD
Location: VA Greater Los Angeles Healthcare System
Neoadjuvant Therapy With Docetaxel and Ketoconazole in Patients With High-Risk Prostate Cancer: A Pilot Study (IST 16167)
Eligible patients with high-risk prostate cancer who are scheduled to undergo radical prostatectomy will receive 4 cycles of therapy with ketoconazole and docetaxel prior to surgery resection. A cycle of therapy is defined as 21 days (3 weeks). Pharmacokinetic analysis will be performed during the first and second cycles of therapy. All patients will be evaluated for toxicity, tumor response, and recurrence.
ID: NCT00870714
Sponsor, Collaborator: Kansas City VAMC; Sanofi
Location: Kansas City VAMC
A Study of Epirubicin With Estramustine Phosphate and Celecoxib for the Treatment of Prostate Cancer
The purpose of this clinical trial is to find out the effect of epirubicin with estramustine phosphate and celecoxib on PSA and objective response in patients with hormone-resistant prostate cancer, as well as to evaluate the toxicity and quality of life of this combination. Celecoxib is an FDA-approved drug that treats arthritis. Epirubicin, alone or with estramustine phosphate, has been used in the treatment of hormone-resistant prostate cancer. These drugs have demonstrated evidence of tumor blood vessel suppression and a combination of these 3 drugs could possibly arrest further tumor growth or even make the tumor decrease in size.
ID: NCT00218205
Sponsor, Collaborator; Investigator: VA New Jersey Health Care System; Pfizer; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Phase II Trial of Combination Therapy With Celecoxib and Taxotere for the Treatment of Stage D3 Prostate Cancer
The purpose of this clinical trial is to find out the safety and effectiveness as well as the patient’s quality of life while taking the combination of Taxotere and celecoxib on patients with hormone refractory prostate cancer. Celecoxib (Celebrex) is an FDA-approved drug that treats arthritis. Taxotere (Docetaxel) is an FDA-approved chemotherapy drug to treat certain forms of cancer. Both drugs have demonstrated evidences of tumor blood vessel suppression and combination of these 2 drugs could possibly arrest further tumor growth or make the tumor decrease in size.
ID: NCT00215345
Sponsor, Collaborator; Investigator: Department of Veterans Affairs, New Jersey; Pfizer, Sanofi; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Yoga Program for Patients Undergoing Prostate Cancer Surgery
Men with localized prostate cancer are often treated with surgery, a treatment that is associated with high rates of adverse effects such as erectile dysfunction (ED) and urinary incontinence (UI) which impact quality of life. Yoga may improve control of UI and improve ED by bringing awareness to and strengthening the pelvic floor musculature. The randomized controlled pilot study is to assess the feasibility of an innovative hybrid (in-person and virtual) twice-weekly yoga program that includes a prehabilitation component and to obtain preliminary data that will help assess its potential effectiveness in alleviating prostate cancer treatment symptom burden (primarily ED and UI). The long-term goal is to develop a scalable and sustainable yoga program that helps cancer survivors manage their treatment side effects.
ID: NCT05929300
Sponsor, Investigator: VA Office of Research and Development; Abigail Silva, PhD, MPH
Location: Edward Hines Jr. VA Hospital
The clinical trials listed below are all open as of July 12, 2024; have ≥ 1 US Department of Veterans Affairs (VA) medical center (VAMC) or US Department of Defense (DoD) military treatment facility location recruiting patients; and are focused on treatments for prostate cancer. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Actively Recruiting
Patient Decision Making About Precision Oncology in Veterans With Advanced Prostate Cancer
This clinical trial explores and implements methods to improve informed decision making regarding precision oncology tests among veterans with prostate cancer that may have spread from where it first started to nearby tissue, lymph nodes, or distant parts of the body (advanced). Precision oncology, the use of germline genetic testing and tumor-based molecular assays to inform cancer care, has become an important aspect of evidence-based care for men with advanced prostate cancer. Veterans with metastatic castrate-resistant prostate cancer may not be carrying out informed decision making due to unmet decisional needs. An informed decision is a choice based on complete and accurate information. The information gained from this study will help researchers develop a decision support intervention and implement the intervention. A decision support intervention may serve as a valuable tool to reduce ongoing racial disparities in genetic testing and encourage enrollment to precision oncology trials.
ID: NCT05396872
Sponsor; Collaborator: University of California, San Francisco; DoD
Location: San Francisco VAMC
DeADT - Living Well With Prostate Cancer
The goal of this pilot randomized implementation trial is to compare 2 strategies to reduce low-value androgen deprivation therapy (ADT) use for prostate cancer care. The aim of the study is to compare implementation of the 2 strategies: use of a clinical reminder order check intervention vs a clinician script/patient education approach, and their impacts on low-value ADT use after 6 months. The main goal of both interventions will be to decrease ADT overuse for patients with prostate cancer, but to do this in a way that is acceptable to the provider who treat these patients. Provider participants will engage with 1 of the interventions triggered in the electronic health record when their patients are deemed likely to receive low-value ADT. Provider participants receive only 1 intervention. The intervention is triggered for every clinic visit involving a patient deemed to be receiving low-value ADT, so provider participants may receive their assigned intervention multiple times. Researchers will compare provider use of both strategies to determine implementation outcomes and whether 1 was more effective in reducing low-value ADT use.
ID: NCT06199986
Sponsor; Collaborator: University of Michigan; VA, National Cancer Institute
Location: VA Ann Arbor Healthcare System
VA Seamless Phase II/III Randomized Trial of Standard Systemic Therapy With or Without PET-Directed Local Therapy for Oligometastatic Prostate Cancer (VA STARPORT)
This is a prospective, open-label, multicenter, seamless phase II to phase III randomized clinical trial designed to compare somatostatin with or without positron emission tomography (PET)-directed local therapy in improving the castration-resistant prostate cancer-free survival for veterans with oligometastatic prostate cancer. Oligometastasis will be defined as 1 to 10 sites of metastatic disease based on the clinical determination.
ID: NCT04787744
Sponsor; Investigators: VA Office of Research and Development; Abhishek Solani, MD, MS, Edward Hines Jr.
Locations: VA Long Beach Healthcare System, VA Greater Los Angeles Healthcare System, Bay Pines VA Healthcare System, Edward Hines Jr. VA Hospital, Richard L. Roudebush VAMC, Baltimore VAMC, VA Boston Healthcare System, VA Ann Arbor Healthcare System, Minneapolis VA Health Care System, Kansas City VAMC, VA New Jersey Healthcare System, VA NY Harbor Healthcare System, Durham VAMC, Louis Stokes VAMC, Corporal Michael J. Crescenz VAMC, Michael E. DeBakey VAMC, Hunter Holmes McGuire VAMC, William S. Middleton Memorial Veterans Hospital, Clement J. Zablocki VAMC
The Prostate Cancer, Genetic Risk, and Equitable Screening Study (ProGRESS)
Prostate cancer is the most common non-skin cancer among veterans and the second leading cause of male cancer death. Current methods of screening men for prostate cancer are inaccurate and cannot identify which men do not have prostate cancer or have low-grade cases that will not cause harm and which men have significant prostate cancer needing treatment. False-positive screening tests can result in unnecessary prostate biopsies for men who do not need them. However, new genetic testing might help identify which men are at highest risk for prostate cancer. This study will examine whether a genetic test helps identify men at risk for significant prostate cancer while helping men who are at low risk for prostate cancer avoid unnecessary biopsies. If this genetic test proves beneficial, it will improve the way that health care providers screen male veterans for prostate cancer.
ID: NCT05926102
Sponsor; Investigator: VA Office of Research and Development; Jason L. Vassy, MD, MPH
Location: VA Boston Healthcare System
Prostate Active Surveillance Study (PASS)
This research study is for men who have chosen active surveillance as a management plan for their prostate cancer. Active surveillance is defined as close monitoring of prostate cancer with the offer of treatment if there are changes in test results. This study seeks to discover markers that will identify cancers that are more aggressive from those tumors that grow slowly.
ID: NCT00756665
Sponsor; Collaborators: University of Washington; Canary Foundation, Early Detection Research Network
Locations: VA San Francisco Health Care System, VA Puget Sound Health Care System
A Study of Checkpoint Inhibitors in Men With Progressive Metastatic Castrate Resistant Prostate Cancer Characterized by a Mismatch Repair Deficiency or Biallelic CDK12 Inactivation (CHOMP)
The primary objective is to assess the activity and efficacy of pembrolizumab, a checkpoint inhibitor, in veterans with metastatic castration-resistant prostate cancer characterized by either mismatch repair deficiency (dMMR) or biallelic inactivation of CDK12 (CDK12-/-). The secondary objectives involve determining the frequency with which dMMR and CDK12-/- occur in this patient population, as well as the effects of pembrolizumab on various clinical endpoints (time to prostate-specific antigen progression, maximal prostate-specific antigen response, time to initiation of alternative antineoplastic therapy, time to radiographic progression, overall survival, and safety and tolerability). Lastly, the study will compare the pretreatment and at-progression metastatic tumor biopsies to investigate the molecular correlates of resistance and sensitivity to pembrolizumab via RNA-sequencing, exome-sequencing, selected protein analyses, and multiplexed immunofluorescence.
ID: NCT04104893
Sponsor; Collaborator: VA Office of Research and Development; Merck Sharp & Dohme LLC
Locations: San Francisco VAMC, VA Greater Los Angeles Healthcare System, Washington DC VAMC, Bay Pines VA Healthcare System Jesse Brown VAMC, VA Ann Arbor Healthcare System, James J. Peter VAMC, VA NY Harbor Healthcare System, Durham VAMC, Corporal Michael J. Crescenz VAMC, Hunter Holmes McGuire VAMC, VA Puget Sound Health Care System
A Single-Arm Phase II Study of Neoadjuvant Intensified Androgen Deprivation (Leuprolideand Abiraterone Acetate) in Combination With AKT Inhibition (Capivasertib) for High-Risk Localized Prostate Cancer With PTEN Loss (SNARE)
The purpose of this study is to learn about how an investigational drug intervention completed before doing prostate surgery (specifically, radical prostatectomy with lymph node dissection) may help in the treatment of high-risk localized prostate cancers that are most resistant to standard treatments. This is a phase II research study. For this study, capivasertib, the study drug, will be taken with intensified androgen deprivation therapy drugs (iADT; abiraterone and leuprolide) prior to radical prostatectomy. This study drug treatment will be evaluated to see if it is effective in shrinking and destroying prostate cancer tumors prior to surgery and to further evaluate its safety prior to prostate cancer surgery.
ID: NCT05593497
Sponsor; Investigator: VA Office of Research and Development; Ryan P. Kopp, MD
Locations: VA Greater Los Angeles Healthcare System, James J. Peters VAMC, VA Portland Health Care System, South Texas Veterans Health Care System, VA Puget Sound Health Care System
Active, Not Recruiting
Intramuscular Mechanisms of Androgen Deprivation-related Sarcopenia
Prostate cancer is the most common cancer among men and is even more common in the military and veteran population. For patients with advanced prostate cancer, the most common treatment includes androgen deprivation therapy (ADT), or the lowering of the levels of the hormone testosterone as much as possible. Unfortunately, ADT also causes patients to be fatigued, weak, and to lose muscle. This is often referred to as “sarcopenia,” and it leads to falls, poor quality of life, and higher risk of death. Currently, there is no treatment for sarcopenia because the investigators do not understand the mechanisms that cause it. The mitochondria are part of the cells responsible for providing energy to muscles, but to this date, the investigators do not know if it is affected in prostate cancer patients with sarcopenia due to ADT.
ID: NCT03867357
Sponsor; Collaborators: Seattle Institute for Biomedical and Clinical Research; DoD, University of Washington
Location: VA Puget Sound Health Care System
Radiation Therapy With or Without Androgen-Deprivation Therapy in Treating Patients With Prostate Cancer
RATIONALE: Radiation therapy uses high-energy X-rays and other types of radiation to kill tumor cells and shrink tumors. Androgens can cause the growth of prostate cancer cells. Androgen deprivation therapy (ADT) may lessen the amount of androgens made by the body. It is not yet known whether radiation therapy is more effective with or without ADT in treating patients with prostate cancer.
PURPOSE: This randomized phase III trial is studying radiation therapy to see how well it works compared with radiation therapy given together with ADT in treating patients with prostate cancer.
ID: NCT00936390
Sponsor; Collaborators: Radiation Therapy Oncology Group; National Cancer Institute, NRG Oncology
Locations: 518 locations, James A. Haley VA Hospital
Enzalutamide With or Without Abiraterone and Prednisone in Treating Patients With Castration-Resistant Metastatic Prostate Cancer
This randomized phase III trial studies enzalutamide to see how well it works compared to enzalutamide, abiraterone, and prednisone in treating patients with castration-resistant metastatic prostate cancer. Androgens can cause the growth of prostate cancer cells. Drugs, such as enzalutamide, abiraterone acetate, and prednisone, may lessen the amount of androgens made by the body.
ID: NCT01949337
Sponsor; Collaborators: Alliance for Clinical Trials in Oncology; NCI, Astellas Pharma US, Inc., Medivation, Inc., Biologics, Inc.
Locations: 539 locations, including VA Connecticut Healthcare System
S1216, Phase III ADT+TAK-700 vs ADT+Bicalutamide for Metastatic Prostate Cancer (S1216)
The purpose of this study is to compare overall survival in newly diagnosed metastatic prostate cancer patients randomly assigned to ADT + TAK-700 vs androgen deprivation therapy (ADT) + bicalutamide.
ID: NCT01809691
Sponsor; Collaborators: SWOG Cancer Research Network; Millennium Pharmaceuticals, Inc., NCI
Locations: 560 locations, including VA New York Harbor Healthcare System
Androgen Ablation Therapy With or Without Chemotherapy in Treating Patients With Metastatic Prostate Cancer (CHAARTED)
RATIONALE: Androgens can cause the growth of prostate cancer cells. Androgen ablation therapy may stop the adrenal glands from making androgens. Drugs used in chemotherapy, such as docetaxel, work in different ways to stop the growth of tumor cells, either by killing the cells or by stopping them from dividing. It is not yet known whether androgen-ablation therapy is more effective with or without docetaxel in treating metastatic prostate cancer.
PURPOSE: This randomized phase III trial is studying androgen ablation therapy and chemotherapy to see how well they work compared to androgen ablation therapy alone in treating patients with metastatic prostate cancer.
ID: NCT00309985
Sponsor; Collaborator: ECOG-ACRIN Cancer Research Group; NCI
Locations: 343 locations, including Mather VAMC
Not Yet Recruiting
biraterone, Enzalutamide, or Apalutamide in Castrate-Sensitive Prostate Cancer
The investigators have used national Veterans Health Administration (VHA) data to demonstrate real-world efficacy of abiraterone and enzalutamide in veterans with metastatic castration-resistant prostate cancer. In the real world that is the VHA, the investigators have successfully estimated g values that accurately predict overall survival, and the use of this metric in other settings should now be explored. In the egalitarian system that is the VHA, the treatment of prostate cancer is excellent, uniform across the US and indifferent to race. The choices made are clearly personalized, given not all men received all therapies, and younger veterans were treated more aggressively.
ID: NCT05422911
Sponsor: James J. Peters VAMC
Location: James J. Peters VAMC
18F-DCFPyL PET/CT Impact on Treatment Strategies for Patients With Prostate Cancer (PROSPYL)
The main purpose of this phase II trial study is to determine whether a positron emission tomography (PET)/computed tomography (CT) scan using 18F-DCFPyL affects the clinical management plan in veterans. In this study, the management plan prior to and after 18F-DCFPyL PET/CT will be recorded by specific questionnaires, and corresponding changes in management will be analyzed. The scan will be used to see how the disease has spread. Both the treatment strategies and probable disease outcomes as relevant to clinical endpoints will be assessed. This study is open to veterans only.
ID: NCT04390880
Sponsor, Investigator: VA Greater Los Angeles Healthcare System; Gholam Berenji, MD
Location: VA Greater Los Angeles Healthcare System
18F-DCFPyL PET-CT Scan and Prostate Cancer
The primary objective of this study is to assess the efficacy of 18F-DCFPyL PET-CT for initial staging of prostate cancer in veterans compared to conventional imaging (99mTc-MDP bone scan and diagnostic CT or MRI). The primary clinical endpoint of our study is the percentage of veterans with prostate cancer in which the 18F-DCFPyL PET-CT identifies M1 disease at initial staging. Secondary objectives included frequency of the change in primary treatment plan after initial staging.
ID: NCT03852654
Sponsor, Investigator: Lida Jafari, MD
Location: VA Greater Los Angeles Healthcare System
Neoadjuvant Therapy With Docetaxel and Ketoconazole in Patients With High-Risk Prostate Cancer: A Pilot Study (IST 16167)
Eligible patients with high-risk prostate cancer who are scheduled to undergo radical prostatectomy will receive 4 cycles of therapy with ketoconazole and docetaxel prior to surgery resection. A cycle of therapy is defined as 21 days (3 weeks). Pharmacokinetic analysis will be performed during the first and second cycles of therapy. All patients will be evaluated for toxicity, tumor response, and recurrence.
ID: NCT00870714
Sponsor, Collaborator: Kansas City VAMC; Sanofi
Location: Kansas City VAMC
A Study of Epirubicin With Estramustine Phosphate and Celecoxib for the Treatment of Prostate Cancer
The purpose of this clinical trial is to find out the effect of epirubicin with estramustine phosphate and celecoxib on PSA and objective response in patients with hormone-resistant prostate cancer, as well as to evaluate the toxicity and quality of life of this combination. Celecoxib is an FDA-approved drug that treats arthritis. Epirubicin, alone or with estramustine phosphate, has been used in the treatment of hormone-resistant prostate cancer. These drugs have demonstrated evidence of tumor blood vessel suppression and a combination of these 3 drugs could possibly arrest further tumor growth or even make the tumor decrease in size.
ID: NCT00218205
Sponsor, Collaborator; Investigator: VA New Jersey Health Care System; Pfizer; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Phase II Trial of Combination Therapy With Celecoxib and Taxotere for the Treatment of Stage D3 Prostate Cancer
The purpose of this clinical trial is to find out the safety and effectiveness as well as the patient’s quality of life while taking the combination of Taxotere and celecoxib on patients with hormone refractory prostate cancer. Celecoxib (Celebrex) is an FDA-approved drug that treats arthritis. Taxotere (Docetaxel) is an FDA-approved chemotherapy drug to treat certain forms of cancer. Both drugs have demonstrated evidences of tumor blood vessel suppression and combination of these 2 drugs could possibly arrest further tumor growth or make the tumor decrease in size.
ID: NCT00215345
Sponsor, Collaborator; Investigator: Department of Veterans Affairs, New Jersey; Pfizer, Sanofi; Basil Kasimis, MD
Location: VA New Jersey Health Care System
A Yoga Program for Patients Undergoing Prostate Cancer Surgery
Men with localized prostate cancer are often treated with surgery, a treatment that is associated with high rates of adverse effects such as erectile dysfunction (ED) and urinary incontinence (UI) which impact quality of life. Yoga may improve control of UI and improve ED by bringing awareness to and strengthening the pelvic floor musculature. The randomized controlled pilot study is to assess the feasibility of an innovative hybrid (in-person and virtual) twice-weekly yoga program that includes a prehabilitation component and to obtain preliminary data that will help assess its potential effectiveness in alleviating prostate cancer treatment symptom burden (primarily ED and UI). The long-term goal is to develop a scalable and sustainable yoga program that helps cancer survivors manage their treatment side effects.
ID: NCT05929300
Sponsor, Investigator: VA Office of Research and Development; Abigail Silva, PhD, MPH
Location: Edward Hines Jr. VA Hospital
The Future of Progressive Multiple Sclerosis Therapies (FULL)
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system, with recent estimates of around 1 million people living with MS in the US.1 In many countries, MS is a leading cause of disability among young adults, second only to trauma.2 Clinically, neurologic worsening (ie, disability) in MS can occur in the relapsing-remitting (RRMS) phase of disease due to incomplete recovery from neuroinflammatory relapses. However, in the 15% of patients with a progressive course from onset (PPMS), and in those with RRMS who transition to a secondary progressive phenotype (SPMS), neurologic worsening follows a slowly progressive pattern.3 A progressive disease course—either PPMS at onset or transitioning to SPMS—is the dominant factor affecting MS-related neurologic disability accumulation. In particular, epidemiologic studies have shown that, in the absence of transitioning to a progressive disease course, < 5% of individuals with MS will accumulate sufficient disability to necessitate use of a cane for ambulation.4-6 Therefore, developing disease modifying therapies (DMTs) that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS represent a critical unmet need.
Research into the development of DMTs for progressive MS has been hindered by a number of factors. In particular, the clinical definition and diagnosis of progressive MS has been an evolving concept. Diagnostic criteria for MS, which help facilitate the enrollment of appropriate subjects into clinical trials, have only recently clarified the current consensus definition for progressive MS—steadily increasing neurologic disability independent of clinical relapses. Looking back to the Schumacher criteria in 1965 and Poser criteria in 1983, it was acknowledged that neurologic symptoms in MS may follow a relapsing-remitting or progressive pattern, but little attempt was made to define progressive MS.7,8 The original McDonald criteria in 2001 defined diagnostic criteria for progressive MS.9 These criteria continued to evolve through subsequent revisions (eg, cerebrospinal fluid [CSF] specific oligoclonal bands no longer are an absolute requirement), and only in the 2017 revision was it emphasized that disability progression must occur independent of clinical relapses, concordant with similar emphasis in the 2013 revision of MS clinical course definitions.3,10
The interpretation of prior clinical trials of DMT for progressive MS must consider this evolving clinical definition. The US Food and Drug Administration (FDA) approved mitoxantrone in 2000—making it the first DMT to carry an approved label for SPMS. While achieving significant clinical efficacy, it is clear from the details of the trial that the enrolled subjects had a high degree of inflammatory disease activity, which suggests that mitoxantrone treats neuroinflammation and not relapse-independent worsening. More recently, disparate results were seen in the anti-CD20 (rituximab, ocrelizumab) and S1P receptor modulator (fingolimod, siponimod) trials targeted at patients with primary and secondary progressive MS.11-14 Although there are differences between these therapies, they are more similar than not within the same therapeutic class. Taken together, these trials illustrate the critical impact the narrower inclusion/exclusion criteria (namely age and extent of inflammatory activity) had on attaining positive outcomes. Other considerations, such as confounding illness, also may impact trial recruitment and generalizability of findings.
The lack of known biological targets in progressive MS, which is a complex disease with an incompletely understood and heterogeneous pathology, also hinders DMT development. Decades of research has characterized multifocal central nervous system (CNS) lesions that exhibit features of demyelination, inflammation, reactive gliosis, axonal loss, and neuronal damage. Until recently, however, much of this research focused on the relapsing phase of disease, and so the understanding of the pathologic underpinnings of progressive disease has remained limited. Current areas of investigation encompass a broad range of pathological processes, such as microglial activation, meningeal lymphoid follicles, remyelination failure, vulnerability of chronically demyelinated axons, oxidative injury, iron accumulation, mitochondrial damage, and others. There is the added complication that the pathologic processes underlying progressive MS are superimposed on the CNS aging process. In particular, the transition to progressive MS and the rate of disability accumulation during progressive MS show strong correlation with age.6,15-17
Finally, DMT development for progressive MS also is hindered by the lack of specific surrogate and clinical outcome measures. Trials for relapsing MS have benefited greatly from the relatively straightforward assessment of clinical relapses and inflammatory disease activity on magnetic resonance imaging (MRI). With the goal of developing DMTs that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS, which by definition occurs independent of clinical relapses, these measures are not directly relevant. The longitudinal clinical disability outcome measures change at a slower rate than in early, relapsing disease. The use of standardized scales (eg, the Expanded Disability Status Scale [EDSS]), lower limb function, upper limb function, cognition, or a combination is a subject of ongoing debate. For example, the ASCEND and IMPACT trials (placebo-controlled trials for SPMS with natalizumab and interferon β-1a, respectively) showed no significant impact in EDSS progression—but in both of these trials, the 9-hole peg test (9-HPT), a performance measure for upper limb function, showed significant improvement.10,18 Particularly in those with an EDSS of > 6.5, who are unlikely to have measurable EDSS progression, functional tests such as the 9-HPT or timed 25-foot walk may be more sensitive as measures for disability progression.11 MRI measures of brain atrophy is the current gold standard surrogate outcome for clinical trials in progressive MS, but others that may warrant consideration include optical coherence tomography (OCT) or CSF markers of axonal degeneration.
DMT for Progressive MS
Current diagnostic nomenclature separates patients with active (superimposed clinical and/or radiographic relapses) from those with inactive disease.3,12 Relapsing forms of MS include all RRMS and those with SPMS with superimposed relapses (ie, active SPMS). Following this paradigm shift, the FDA changed the indication for already approved DMT from RRMS to relapsing forms of MS. Below is a discussion of DMT that specifically use the term SPMS and PPMS in the indication, where phase 3 trial data for progressive MS is available.
In 2019, the FDA approved the first oral medication (siponimod) for active SPMS. Subsequently, updates to the labels of the older DMT expanded to include active SPMS. Until 2019, the only FDA approved medication for SPMS was mitoxantrone, and use of this medication was limited due to unfavorable adverse effects (AEs). No medications had obtained FDA approval for inactive SPMS to this point, which represented an unmet need for a considerable number of patients.
Mitoxantrone became the first DMT approved for use in SPMS in 2000 following early trials that showed reductions in EDSS worsening, change in ambulation index, reduced number of treated relapses, and prolonged time to first treated relapse. However, as with some of the other positive trials in progressive MS, it is difficult to discern the impact of suppression of relapses as opposed to direct impact on progressive pathophysiology. Within the placebo arm, for example, there were 129 relapses among the 64 subjects, which suggests that these cases had particularly active disease or were in the early stages of SPMS.13 Furthermore, the use of this medication was limited due to concerns of cardiotoxicity and hematologic malignancy as serious AEs.
The trials of interferon β-1b illustrate the same difficulty of isolating possible benefits in disease progression from disability accumulated from relapses. The first interferon β-1b trial for SPMS, was conducted in Europe using fingolimod and showed a delay in confirmed disability progression compared to placebo as measured with the EDSS.14 However, a North American trial that followed in 2004 was unable to replicate this finding.15 The patients in the European trial appeared to be in an earlier phase of SPMS with more active disease, and a post-hoc pooled analysis suggested that patients with active disease and those with more pronounced disability progression were more likely to benefit from treatment.16 Overall, interferons do not appear to appreciably alter disability in the long-term for patients with SPMS, though they may modify short-term, relapse-related disability.
Perhaps the most encouraging data for SPMS was found in the EXPAND trial, which investigated siponimod, an S1P receptor modulator that is more selective than fingolimod. The trial identified a 21% reduction in 3-month confirmed disability progression for SPMS patients taking siponimod compared with those taking a placebo.17 Although the patients in EXPAND did seem to have relatively less disease activity at baseline than those who participated in other SPMS trials, those who benefitted from siponimod were primarily patients who had clinical and/or radiographic relapses over the prior 2 years. Based on this, the FDA approved siponimod for active SPMS. The extent to which siponimod exerts a true neuroprotective effect beyond reducing inflammation has not been clearly established.
B-cell depleting therapies rituximab and ocrelizumab have been evaluated in both primary and secondary progressive MS populations. Early investigations of the chimeric rituximab in PPMS did not show benefits on disability (EDSS) progression; however, benefits were seen in analysis of some subgroups.18 With this in mind, the ORATORIO trial for the humanized version, ocreluzimab, included PPMS patients that were younger (aged < 55 years) and had cutoffs for disease duration (< 15 years for those with EDSS more than 5 years, < 10 years for those with EDSS less than 5 years). The study showed statistically significant changes on disability progression, which led to ocrelizumab receiving the first FDA indication for PPMS.11 There are substantial pathophysiologic similarities between PPMS and SPMS in the progressive phase.19 While these medications may have similar effects across these disease processes, these benefits have not yet been demonstrated in a prospective trial for the SPMS population. Regardless, B-cell depleting therapy is a reasonable consideration for select patients with active SPMS, consistent with a relapsing form of MS.
Therapies in Development
DMT development for progressive MS is a high priority area. Current immunomodulatory therapies for RRMS have consistently been ineffective in the inactive forms of MS, with the possible exceptions of ocrelizumab and siponimod. Therefore, instead of immunosuppression, many agents currently in phase 2 and 3 clinical trials target alternative pathophysiological processes in order to provide neuroprotection, and/or promote remyelination and neurogenesis. These targets include oxidative stress (OS), non-T cell mediated inflammation, and mitochondrial/energy failure.20 Below we review a selection of clinical trials testing agents following these approaches. Many agents have more than one potential mechanism of action (MOA) that could benefit progressive MS.
Lipoic acid (LA), also known as α-lipoic acid and thiotic acid, is one such agent targeting alternative pathophysiology in SPMS. LA is an endogenous agent synthesized de novo from fatty acids and cysteine as well as obtained in small amounts from foods.21 It has antioxidant (AO) properties including direct radical scavenging, regeneration of glutathione, and upregulation of AO enzymes via the NrF2 pathway.22 It supports mitochondria as a key cofactor for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, and it also aids mitochondrial DNA synthesis.21,22 Studies in experimental autoimmune encephalomyelitis, a widely used experimental mouse model of inflammatory demyelinating disease, also indicate a reduction in excessive microglial activation.23 A phase 2 pilot randomized controlled trial (RCT) of 1200 mg LA in SPMS (n = 51) resulted in significantly less whole brain atrophy by SIENA (Structural Image Evaluation, Using Normalization, of Atrophy) at 2 years.24 A follow-up multicenter trial is ongoing.
Simvastatin also targets alternative pathophysiology in SPMS. It has anti-inflammatory effects, improves vascular function, and promotes neuroprotection by reducing excitotoxicity. A phase 2 RCT demonstrated a reduction in whole brain atrophy in SPMS (n = 140), and a phase 3 trial is underway.25 Ibudilast is another repurposed drug that targets alternative inflammation by inhibiting several cyclic nucleotide phosphodiesterases, macrophage migration inhibitory factor and toll-like receptor 4. A phase 2 trial (n = 225) in both SPMS and PPMS also demonstrated a reduction in brain atrophy, but participants had high rates of AEs.26
Lithium and riluzole promote neuroprotection by reducing excitotoxicity. Lithium is a pharmacologic active cation used as a mood stabilizer and causes inhibition of glycogen synthase kinase-3β. Animal models also indicate that lithium may decrease inflammation and positively impact neurogenesis.27 A crossover pilot trial demonstrated tolerability with trends toward stabilization of EDSS and reductions in brain atrophy.28 Three neuroprotective agents, riluzole (reduces glutamate excitotoxicity), fluoxetine (stimulates glycogenolysis and improves mitochondrial energy production), and amiloride (an acid-sensing ion channel blocker that opens in response to inflammation) were tested in a phase 2b multi-arm, multi-site parallel group RCT in SPMS (n = 445). The study failed to yield differences from placebo for any agent in reduction of brain volume loss.29 A prior study of lamotrigine, a sodium channel blocker, also failed to find changes in brain volume loss.30 These studies highlight the large sample sizes and/or long study durations needed to test agents using brain atrophy as primary outcome. In the future, precise surrogate markers of neuroprotection will be a great need for earlier phase trials. These results also suggest that targeting > 1 MOA may be necessary to treat SPMS effectively.
Efforts to promote remyelination target one hallmark of MS damage. High dose biotin (about 10,000× usual dose) may promote myelin repair as a cofactor for fatty acid synthesis and support mitochondrial oxidative phosphorylation. While a RCT yielded a greater proportion of participants with either PPMS or SPMS with improvement in disability than placebo at 12 months, an open label trial suggested otherwise indicating a need for a more definitive trial.31,32
Anti-LINGO-1 (opicinumab) is a monoclonal antibody that targets LINGO, a potent negative regulator of oligodendrocyte differentiation and myelination.33 Although this agent failed in a phase 2 trial in relapsing MS, and is thus unlikely to be tested in progressive forms, the innovative approach to stimulating oligodendrocytes is ongoing. One such effort is to use thyroid hormone, crucial to myelin formation during development, as a repair agent in MS.34 A dose-finding study of thyroid hormone was completed and efforts to develop a thyromimetic agent are ongoing.
Finally, efforts to promote neurogenesis remain a goal for many neurodegenerative diseases. Exercise appears to prevent age-related atrophy of the hippocampus in animals and humans and help maintain neuronal health.35 In patients with RRMS, cortical thickness is impacted positively by resistance training, which suggests a neuroprotective effect.36 A multi-center trial of exercise in patients with progressive MS investigating cognitive outcomes is ongoing.
Discontinuing DMT
In the early 1990s, the successful development of immune modulating therapies that reliably reduced disease activity in RRMS led to widespread initiation in patients with relapsing disease. However, guidance on when or if to discontinue DMT, even in those who have transitioned to SPMS, remains largely absent at this time. Requests to discontinue DMT may come from patients weary of taking medication (especially injections), bothered by AEs, or those who no longer perceive efficacy from their treatments. Clinicians also may question the benefit of immune modulation in patients with longstanding freedom from relapses or changes in MRI lesion burden.
To inform discussion centered on treatment discontinuation, a clinical trial is currently underway to better answer the question of when and how to withdraw MS therapy. Discontinuation of Disease Modifying Therapies in Multiple Sclerosis (DISCO-MS) is a prospective, placebo-controlled RCT and its primary endpoint is recurrence of disease activity over a 2 year follow-up period.37 Eligibility requirements for the trial include age > 55 years, 5-year freedom from relapses, and 3-year freedom from new MRI lesions (criteria informed by progressive MS cohort studies).31 In addition to demonstrating the active disease recurrence rates in this patient population, the trial also aims to identify risk factors for recurrent disease activity among treated MS patients.37 DISCO-MS builds upon a series of retrospective and observational studies that partially answered these questions, albeit in the context of biases inherent in retrospective or observational studies.
A Minneapolis MS Treatment and Research Center single-center study identified 77 SPMS patients with no acute CNS inflammatory events over 2 to 20 years and advised these patients to stop taking DMT.32 In this group, 11.7% of subjects experienced recurrent active disease. Age was the primary discriminating factor. The mean age of those who experienced disease activity was 56 years vs 61 years those who did not. A second observational study from France found that among 100 SPMS patients treated either with interferon β or glatiramer acetate for at least 6 months, 35% experienced some form of inflammatory disease upon discontinuation.38 Sixteen patients relapsed and 19 developed gadolinium-enhancing MR lesions after DMT discontinuation. However, the age of the cohort was younger than the Minneapolis study (47.2 years vs 61 years), and reasons for discontinuation (eg, AEs or lack of disease activity) were not considered in the analysis.
Other studies examining the safety of DMT discontinuation have not considered MS subtype or excluded patients with progressive subtypes of MS. The largest studies to date on DMT discontinuation utilized the international MSBase global patient registry, which identified nearly 5,000 patients who discontinued interferons (73%), glatiramer acetate (18%), natalizumab (6%), or fingolimod (3%), without specifying the reasons for discontinuation.39 Despite these shortcomings, data reveal trends that are helpful in predicting how MS tends to behave in patients who have discontinued therapy. Not surprisingly, disability progression was most likely among patients already characterized as having a progressive phenotype, while relapses were less likely to occur among older, progressive patients.
Although clinicians may be increasingly willing to discuss DMT discontinuation with their patients, at least 1 study exploring patient perspectives on stopping treatment suggests widespread reluctance to stop treatment. A survey conducted with participants in the North American Research Committee on Multiple Sclerosis patient-report registry found that among survey respondents, only 11.9% would discontinue their MS medication if deemed stable, while 66.3% stated they were unlikely to stop treatment.40
These results suggest that before clinicians incorporate DMT discontinuation into the normal course of discussion with patients, they should be prepared to provide both education (on the wisdom of stopping under the right circumstances) and evidence to support when and how to make the recommendation. Based on existing evidence, criteria for recommending treatment discontinuation might include prolonged freedom from disease activity (≥ 5 years), age > 55 years or 60 years, and a progressive disease course. Thus far, no combination of factors has been shown to completely predict an event-free transition off of medicine. Since no fixed algorithm yet exists to guide DMT stoppage in MS, reasonable suggestions for monitoring patients might include surveillance MRIs, more frequent clinic visits, and possible transitional treatment for patients coming off of natalizumab or fingolimod, since these drugs have been associated with rebound disease activity when discontinued.41,42
Clinicians wishing to maximize function and quality of life for their patients at any age or stage of disease should look to nonpharmacologic interventions to lessen disability and maximize quality of life. While beyond the scope of this discussion, preliminary evidence suggests multimodal (aerobic, resistance, balance) exercise may enhance endurance and cognitive processing speed, and that treatment of comorbid diseases affecting vascular health benefits MS. 43
Conclusions
The development of numerous treatments for RRMS has established an entirely new landscape and disease course for those with MS. While this benefit has not entirely extended to those with progressive MS, those with active disease with superimposed relapses may receive limited benefit from these medications. New insights into the pathophysiology of progressive MS may lead us to new treatments through multiple alternative pathophysiologic pathways. Some early studies using this strategy show promise in slowing the progressive phase. Medication development for progressive MS faces multiple challenges due to lack of a single animal model demonstrating both pathology and clinical effects, absence of phase 1 surrogate biomarkers, and later phase trial endpoints that require large sample sizes and extended study durations. Nevertheless, the increase in number of trials and diversity of therapeutic approaches for progressive MS provides hope for effective therapy. Currently, the heterogeneity of the population with progressive MS requires an individualized treatment approach, and in some of these patients, stopping therapy may be a reasonable consideration. Symptomatic management remains critical for all patients with progressive MS as well as non-pharmacologic approaches that maximize quality of life.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data [published correction appears in Neurology. 2019;93(15):688]. Neurology. 2019;92(10):e1029-e1040.
2. Browne P, Chandraratna D, Angood C, et al. Atlas of multiple sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83(11):1022-1024.
3. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
4. Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112(Pt 1):133-146. 5. Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain. 2006;129(Pt 3):595-605.
6. Tutuncu M, Tang J, Zeid NA, et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult Scler. 2013;19(2):188-198.
7. Schumacher GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci. 1965;122:552-568.
8. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13(3):227-231.
9. McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121-127.
10. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
11. Montalban X, Hauser SL, Kappos L, et al; ORATORIO Clinical Investigators. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209-220.
12. Hawker K, O’Connor P, Freedman MS, et al; OLYMPUS trial group. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
13. Kappos L, Bar-Or A, Cree BAC, et al; EXPAND Clinical Investigators. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study [published correction appears in Lancet. 2018;392(10160):2170]. Lancet. 2018;391(10127):1263-1273.
14. Lublin F, Miller DH, Freedman MS, et al; INFORMS study investigators. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial [published correction appears in Lancet. 2017;389(10066):254]. Lancet. 2016;387(10023):1075-1084.
15. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343(20):1430-1438.
16. Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(Pt 3):584-594.
17. Leray E, Yaouanq J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(Pt 7):1900–1913.
18. Kapoor R, Ho PR, Campbell N, et al; ASCEND investigators. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405-415.
19. Koch MW, Mostert J, Uitdehaag B, Cutter G. Clinical outcome measures in SPMS trials: an analysis of the IMPACT and ASCEND original trial data sets [published online ahead of print, 2019 Sep 13]. Mult Scler. 2019;1352458519876701.
20. Hartung HP, Gonsette R, König N, et al; Mitoxantrone in Multiple Sclerosis Study Group (MIMS). Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018-2025.
21. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352(9139):1491-1497.
22. Gorąca A, Huk-Kolega H, Piechota A, Kleniewska P, Ciejka E, Skibska B. Lipoic acid - biological activity and therapeutic potential. Pharmacol Rep. 2011;63:849-858.
23. Chaudhary P, Marracci G, Pocius E, Galipeau D, Morris B, Bourdette D. Effects of lipoic acid on primary murine microglial cells. J Neuroimmunol. 2019;334:576972.
24. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4:e374.
25. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213-2221.
26. Fox RJ, Coffey CS, Conwit R, et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med. 2018;379:846-855.
27. Rinker JR, 2nd, Cossey TC, Cutter GR, Culpepper WJ. A retrospective review of lithium usage in veterans with multiple sclerosis. Mult Scler Relat Disord. 2013;2:327-333.
28. Rinker JR, W Meador, V Sung, A Nicholas, G Cutter. Results of a pilot trial of lithium in progressive multiple sclerosis. ECTRIMS Online Library. 09/16/16; 145965; P12822016.
29. Chataway J, De Angelis F, Connick P, et al; MS-SMART Investigators. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): a phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020;19(3):214-225.
30. Kapoor R, Furby J, Hayton T, et al. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9:681-688.
31. Paz Soldan MM, Novotna M, Abou Zeid N, et al. Relapses and disability accumulation in progressive multiple sclerosis. Neurology. 2015;84:81-88.
32. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19:11-14.
33. Ruggieri S, Tortorella C, Gasperini C. Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev Neurother 2017;17:1081-1089.
34. Hartley MD, Banerji T, Tagge IJ, et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. 2019;4(8):e126329.
35. Firth J, Stubbs B, Vancampfort D, et al. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage. 2018;166:230-238.
36. Kjolhede T, Siemonsen S, Wenzel D, et al. Can resistance training impact MRI outcomes in relapsing-remitting multiple sclerosis? Mult Scler. 2018;24:1356-1365.
37. US National Library of Medicine, Clinicaltrials.gov. Discontinuation of Disease Modifying Therapies (DMTs) in Multiple Sclerosis (MS) (DISCOMS). https://clinicaltrials.gov/ct2/show/NCT03073603. Updated February 10, 2020. Accessed March 26, 2020.
38. Bonenfant J, Bajeux E, Deburghgraeve V, Le Page E, Edan G, Kerbrat A. Can we stop immunomodulatory treatments in secondary progressive multiple sclerosis? Eur J Neurol. 2017;24:237-244.
39. Kister I, Spelman T, Patti F, et al. Predictors of relapse and disability progression in MS patients who discontinue disease-modifying therapy. J Neurol Sci. 2018;391:72-76.
40. McGinley MP, Cola PA, Fox RJ, Cohen JA, Corboy JJ, Miller D. Perspectives of individuals with multiple sclerosis on discontinuation of disease-modifying therapies. Mult Scler. 2019:1352458519867314.
41. Hatcher SE, Waubant E, Graves JS. Rebound Syndrome in Multiple Sclerosis After Fingolimod Cessation-Reply. JAMA Neurol. 2016;73:1376.
42. Vellinga MM, Castelijns JA, Barkhof F, Uitdehaag BM, Polman CH. Postwithdrawal rebound increase in T2 lesional activity in natalizumab-treated MS patients. Neurology. 2008;70:1150-1151.
43. Sandroff BM, Bollaert RE, Pilutti LA, et al. Multimodal exercise training in multiple sclerosis: A randomized controlled trial in persons with substantial mobility disability. Contemp Clin Trials 2017;61:39-47.
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system, with recent estimates of around 1 million people living with MS in the US.1 In many countries, MS is a leading cause of disability among young adults, second only to trauma.2 Clinically, neurologic worsening (ie, disability) in MS can occur in the relapsing-remitting (RRMS) phase of disease due to incomplete recovery from neuroinflammatory relapses. However, in the 15% of patients with a progressive course from onset (PPMS), and in those with RRMS who transition to a secondary progressive phenotype (SPMS), neurologic worsening follows a slowly progressive pattern.3 A progressive disease course—either PPMS at onset or transitioning to SPMS—is the dominant factor affecting MS-related neurologic disability accumulation. In particular, epidemiologic studies have shown that, in the absence of transitioning to a progressive disease course, < 5% of individuals with MS will accumulate sufficient disability to necessitate use of a cane for ambulation.4-6 Therefore, developing disease modifying therapies (DMTs) that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS represent a critical unmet need.
Research into the development of DMTs for progressive MS has been hindered by a number of factors. In particular, the clinical definition and diagnosis of progressive MS has been an evolving concept. Diagnostic criteria for MS, which help facilitate the enrollment of appropriate subjects into clinical trials, have only recently clarified the current consensus definition for progressive MS—steadily increasing neurologic disability independent of clinical relapses. Looking back to the Schumacher criteria in 1965 and Poser criteria in 1983, it was acknowledged that neurologic symptoms in MS may follow a relapsing-remitting or progressive pattern, but little attempt was made to define progressive MS.7,8 The original McDonald criteria in 2001 defined diagnostic criteria for progressive MS.9 These criteria continued to evolve through subsequent revisions (eg, cerebrospinal fluid [CSF] specific oligoclonal bands no longer are an absolute requirement), and only in the 2017 revision was it emphasized that disability progression must occur independent of clinical relapses, concordant with similar emphasis in the 2013 revision of MS clinical course definitions.3,10
The interpretation of prior clinical trials of DMT for progressive MS must consider this evolving clinical definition. The US Food and Drug Administration (FDA) approved mitoxantrone in 2000—making it the first DMT to carry an approved label for SPMS. While achieving significant clinical efficacy, it is clear from the details of the trial that the enrolled subjects had a high degree of inflammatory disease activity, which suggests that mitoxantrone treats neuroinflammation and not relapse-independent worsening. More recently, disparate results were seen in the anti-CD20 (rituximab, ocrelizumab) and S1P receptor modulator (fingolimod, siponimod) trials targeted at patients with primary and secondary progressive MS.11-14 Although there are differences between these therapies, they are more similar than not within the same therapeutic class. Taken together, these trials illustrate the critical impact the narrower inclusion/exclusion criteria (namely age and extent of inflammatory activity) had on attaining positive outcomes. Other considerations, such as confounding illness, also may impact trial recruitment and generalizability of findings.
The lack of known biological targets in progressive MS, which is a complex disease with an incompletely understood and heterogeneous pathology, also hinders DMT development. Decades of research has characterized multifocal central nervous system (CNS) lesions that exhibit features of demyelination, inflammation, reactive gliosis, axonal loss, and neuronal damage. Until recently, however, much of this research focused on the relapsing phase of disease, and so the understanding of the pathologic underpinnings of progressive disease has remained limited. Current areas of investigation encompass a broad range of pathological processes, such as microglial activation, meningeal lymphoid follicles, remyelination failure, vulnerability of chronically demyelinated axons, oxidative injury, iron accumulation, mitochondrial damage, and others. There is the added complication that the pathologic processes underlying progressive MS are superimposed on the CNS aging process. In particular, the transition to progressive MS and the rate of disability accumulation during progressive MS show strong correlation with age.6,15-17
Finally, DMT development for progressive MS also is hindered by the lack of specific surrogate and clinical outcome measures. Trials for relapsing MS have benefited greatly from the relatively straightforward assessment of clinical relapses and inflammatory disease activity on magnetic resonance imaging (MRI). With the goal of developing DMTs that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS, which by definition occurs independent of clinical relapses, these measures are not directly relevant. The longitudinal clinical disability outcome measures change at a slower rate than in early, relapsing disease. The use of standardized scales (eg, the Expanded Disability Status Scale [EDSS]), lower limb function, upper limb function, cognition, or a combination is a subject of ongoing debate. For example, the ASCEND and IMPACT trials (placebo-controlled trials for SPMS with natalizumab and interferon β-1a, respectively) showed no significant impact in EDSS progression—but in both of these trials, the 9-hole peg test (9-HPT), a performance measure for upper limb function, showed significant improvement.10,18 Particularly in those with an EDSS of > 6.5, who are unlikely to have measurable EDSS progression, functional tests such as the 9-HPT or timed 25-foot walk may be more sensitive as measures for disability progression.11 MRI measures of brain atrophy is the current gold standard surrogate outcome for clinical trials in progressive MS, but others that may warrant consideration include optical coherence tomography (OCT) or CSF markers of axonal degeneration.
DMT for Progressive MS
Current diagnostic nomenclature separates patients with active (superimposed clinical and/or radiographic relapses) from those with inactive disease.3,12 Relapsing forms of MS include all RRMS and those with SPMS with superimposed relapses (ie, active SPMS). Following this paradigm shift, the FDA changed the indication for already approved DMT from RRMS to relapsing forms of MS. Below is a discussion of DMT that specifically use the term SPMS and PPMS in the indication, where phase 3 trial data for progressive MS is available.
In 2019, the FDA approved the first oral medication (siponimod) for active SPMS. Subsequently, updates to the labels of the older DMT expanded to include active SPMS. Until 2019, the only FDA approved medication for SPMS was mitoxantrone, and use of this medication was limited due to unfavorable adverse effects (AEs). No medications had obtained FDA approval for inactive SPMS to this point, which represented an unmet need for a considerable number of patients.
Mitoxantrone became the first DMT approved for use in SPMS in 2000 following early trials that showed reductions in EDSS worsening, change in ambulation index, reduced number of treated relapses, and prolonged time to first treated relapse. However, as with some of the other positive trials in progressive MS, it is difficult to discern the impact of suppression of relapses as opposed to direct impact on progressive pathophysiology. Within the placebo arm, for example, there were 129 relapses among the 64 subjects, which suggests that these cases had particularly active disease or were in the early stages of SPMS.13 Furthermore, the use of this medication was limited due to concerns of cardiotoxicity and hematologic malignancy as serious AEs.
The trials of interferon β-1b illustrate the same difficulty of isolating possible benefits in disease progression from disability accumulated from relapses. The first interferon β-1b trial for SPMS, was conducted in Europe using fingolimod and showed a delay in confirmed disability progression compared to placebo as measured with the EDSS.14 However, a North American trial that followed in 2004 was unable to replicate this finding.15 The patients in the European trial appeared to be in an earlier phase of SPMS with more active disease, and a post-hoc pooled analysis suggested that patients with active disease and those with more pronounced disability progression were more likely to benefit from treatment.16 Overall, interferons do not appear to appreciably alter disability in the long-term for patients with SPMS, though they may modify short-term, relapse-related disability.
Perhaps the most encouraging data for SPMS was found in the EXPAND trial, which investigated siponimod, an S1P receptor modulator that is more selective than fingolimod. The trial identified a 21% reduction in 3-month confirmed disability progression for SPMS patients taking siponimod compared with those taking a placebo.17 Although the patients in EXPAND did seem to have relatively less disease activity at baseline than those who participated in other SPMS trials, those who benefitted from siponimod were primarily patients who had clinical and/or radiographic relapses over the prior 2 years. Based on this, the FDA approved siponimod for active SPMS. The extent to which siponimod exerts a true neuroprotective effect beyond reducing inflammation has not been clearly established.
B-cell depleting therapies rituximab and ocrelizumab have been evaluated in both primary and secondary progressive MS populations. Early investigations of the chimeric rituximab in PPMS did not show benefits on disability (EDSS) progression; however, benefits were seen in analysis of some subgroups.18 With this in mind, the ORATORIO trial for the humanized version, ocreluzimab, included PPMS patients that were younger (aged < 55 years) and had cutoffs for disease duration (< 15 years for those with EDSS more than 5 years, < 10 years for those with EDSS less than 5 years). The study showed statistically significant changes on disability progression, which led to ocrelizumab receiving the first FDA indication for PPMS.11 There are substantial pathophysiologic similarities between PPMS and SPMS in the progressive phase.19 While these medications may have similar effects across these disease processes, these benefits have not yet been demonstrated in a prospective trial for the SPMS population. Regardless, B-cell depleting therapy is a reasonable consideration for select patients with active SPMS, consistent with a relapsing form of MS.
Therapies in Development
DMT development for progressive MS is a high priority area. Current immunomodulatory therapies for RRMS have consistently been ineffective in the inactive forms of MS, with the possible exceptions of ocrelizumab and siponimod. Therefore, instead of immunosuppression, many agents currently in phase 2 and 3 clinical trials target alternative pathophysiological processes in order to provide neuroprotection, and/or promote remyelination and neurogenesis. These targets include oxidative stress (OS), non-T cell mediated inflammation, and mitochondrial/energy failure.20 Below we review a selection of clinical trials testing agents following these approaches. Many agents have more than one potential mechanism of action (MOA) that could benefit progressive MS.
Lipoic acid (LA), also known as α-lipoic acid and thiotic acid, is one such agent targeting alternative pathophysiology in SPMS. LA is an endogenous agent synthesized de novo from fatty acids and cysteine as well as obtained in small amounts from foods.21 It has antioxidant (AO) properties including direct radical scavenging, regeneration of glutathione, and upregulation of AO enzymes via the NrF2 pathway.22 It supports mitochondria as a key cofactor for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, and it also aids mitochondrial DNA synthesis.21,22 Studies in experimental autoimmune encephalomyelitis, a widely used experimental mouse model of inflammatory demyelinating disease, also indicate a reduction in excessive microglial activation.23 A phase 2 pilot randomized controlled trial (RCT) of 1200 mg LA in SPMS (n = 51) resulted in significantly less whole brain atrophy by SIENA (Structural Image Evaluation, Using Normalization, of Atrophy) at 2 years.24 A follow-up multicenter trial is ongoing.
Simvastatin also targets alternative pathophysiology in SPMS. It has anti-inflammatory effects, improves vascular function, and promotes neuroprotection by reducing excitotoxicity. A phase 2 RCT demonstrated a reduction in whole brain atrophy in SPMS (n = 140), and a phase 3 trial is underway.25 Ibudilast is another repurposed drug that targets alternative inflammation by inhibiting several cyclic nucleotide phosphodiesterases, macrophage migration inhibitory factor and toll-like receptor 4. A phase 2 trial (n = 225) in both SPMS and PPMS also demonstrated a reduction in brain atrophy, but participants had high rates of AEs.26
Lithium and riluzole promote neuroprotection by reducing excitotoxicity. Lithium is a pharmacologic active cation used as a mood stabilizer and causes inhibition of glycogen synthase kinase-3β. Animal models also indicate that lithium may decrease inflammation and positively impact neurogenesis.27 A crossover pilot trial demonstrated tolerability with trends toward stabilization of EDSS and reductions in brain atrophy.28 Three neuroprotective agents, riluzole (reduces glutamate excitotoxicity), fluoxetine (stimulates glycogenolysis and improves mitochondrial energy production), and amiloride (an acid-sensing ion channel blocker that opens in response to inflammation) were tested in a phase 2b multi-arm, multi-site parallel group RCT in SPMS (n = 445). The study failed to yield differences from placebo for any agent in reduction of brain volume loss.29 A prior study of lamotrigine, a sodium channel blocker, also failed to find changes in brain volume loss.30 These studies highlight the large sample sizes and/or long study durations needed to test agents using brain atrophy as primary outcome. In the future, precise surrogate markers of neuroprotection will be a great need for earlier phase trials. These results also suggest that targeting > 1 MOA may be necessary to treat SPMS effectively.
Efforts to promote remyelination target one hallmark of MS damage. High dose biotin (about 10,000× usual dose) may promote myelin repair as a cofactor for fatty acid synthesis and support mitochondrial oxidative phosphorylation. While a RCT yielded a greater proportion of participants with either PPMS or SPMS with improvement in disability than placebo at 12 months, an open label trial suggested otherwise indicating a need for a more definitive trial.31,32
Anti-LINGO-1 (opicinumab) is a monoclonal antibody that targets LINGO, a potent negative regulator of oligodendrocyte differentiation and myelination.33 Although this agent failed in a phase 2 trial in relapsing MS, and is thus unlikely to be tested in progressive forms, the innovative approach to stimulating oligodendrocytes is ongoing. One such effort is to use thyroid hormone, crucial to myelin formation during development, as a repair agent in MS.34 A dose-finding study of thyroid hormone was completed and efforts to develop a thyromimetic agent are ongoing.
Finally, efforts to promote neurogenesis remain a goal for many neurodegenerative diseases. Exercise appears to prevent age-related atrophy of the hippocampus in animals and humans and help maintain neuronal health.35 In patients with RRMS, cortical thickness is impacted positively by resistance training, which suggests a neuroprotective effect.36 A multi-center trial of exercise in patients with progressive MS investigating cognitive outcomes is ongoing.
Discontinuing DMT
In the early 1990s, the successful development of immune modulating therapies that reliably reduced disease activity in RRMS led to widespread initiation in patients with relapsing disease. However, guidance on when or if to discontinue DMT, even in those who have transitioned to SPMS, remains largely absent at this time. Requests to discontinue DMT may come from patients weary of taking medication (especially injections), bothered by AEs, or those who no longer perceive efficacy from their treatments. Clinicians also may question the benefit of immune modulation in patients with longstanding freedom from relapses or changes in MRI lesion burden.
To inform discussion centered on treatment discontinuation, a clinical trial is currently underway to better answer the question of when and how to withdraw MS therapy. Discontinuation of Disease Modifying Therapies in Multiple Sclerosis (DISCO-MS) is a prospective, placebo-controlled RCT and its primary endpoint is recurrence of disease activity over a 2 year follow-up period.37 Eligibility requirements for the trial include age > 55 years, 5-year freedom from relapses, and 3-year freedom from new MRI lesions (criteria informed by progressive MS cohort studies).31 In addition to demonstrating the active disease recurrence rates in this patient population, the trial also aims to identify risk factors for recurrent disease activity among treated MS patients.37 DISCO-MS builds upon a series of retrospective and observational studies that partially answered these questions, albeit in the context of biases inherent in retrospective or observational studies.
A Minneapolis MS Treatment and Research Center single-center study identified 77 SPMS patients with no acute CNS inflammatory events over 2 to 20 years and advised these patients to stop taking DMT.32 In this group, 11.7% of subjects experienced recurrent active disease. Age was the primary discriminating factor. The mean age of those who experienced disease activity was 56 years vs 61 years those who did not. A second observational study from France found that among 100 SPMS patients treated either with interferon β or glatiramer acetate for at least 6 months, 35% experienced some form of inflammatory disease upon discontinuation.38 Sixteen patients relapsed and 19 developed gadolinium-enhancing MR lesions after DMT discontinuation. However, the age of the cohort was younger than the Minneapolis study (47.2 years vs 61 years), and reasons for discontinuation (eg, AEs or lack of disease activity) were not considered in the analysis.
Other studies examining the safety of DMT discontinuation have not considered MS subtype or excluded patients with progressive subtypes of MS. The largest studies to date on DMT discontinuation utilized the international MSBase global patient registry, which identified nearly 5,000 patients who discontinued interferons (73%), glatiramer acetate (18%), natalizumab (6%), or fingolimod (3%), without specifying the reasons for discontinuation.39 Despite these shortcomings, data reveal trends that are helpful in predicting how MS tends to behave in patients who have discontinued therapy. Not surprisingly, disability progression was most likely among patients already characterized as having a progressive phenotype, while relapses were less likely to occur among older, progressive patients.
Although clinicians may be increasingly willing to discuss DMT discontinuation with their patients, at least 1 study exploring patient perspectives on stopping treatment suggests widespread reluctance to stop treatment. A survey conducted with participants in the North American Research Committee on Multiple Sclerosis patient-report registry found that among survey respondents, only 11.9% would discontinue their MS medication if deemed stable, while 66.3% stated they were unlikely to stop treatment.40
These results suggest that before clinicians incorporate DMT discontinuation into the normal course of discussion with patients, they should be prepared to provide both education (on the wisdom of stopping under the right circumstances) and evidence to support when and how to make the recommendation. Based on existing evidence, criteria for recommending treatment discontinuation might include prolonged freedom from disease activity (≥ 5 years), age > 55 years or 60 years, and a progressive disease course. Thus far, no combination of factors has been shown to completely predict an event-free transition off of medicine. Since no fixed algorithm yet exists to guide DMT stoppage in MS, reasonable suggestions for monitoring patients might include surveillance MRIs, more frequent clinic visits, and possible transitional treatment for patients coming off of natalizumab or fingolimod, since these drugs have been associated with rebound disease activity when discontinued.41,42
Clinicians wishing to maximize function and quality of life for their patients at any age or stage of disease should look to nonpharmacologic interventions to lessen disability and maximize quality of life. While beyond the scope of this discussion, preliminary evidence suggests multimodal (aerobic, resistance, balance) exercise may enhance endurance and cognitive processing speed, and that treatment of comorbid diseases affecting vascular health benefits MS. 43
Conclusions
The development of numerous treatments for RRMS has established an entirely new landscape and disease course for those with MS. While this benefit has not entirely extended to those with progressive MS, those with active disease with superimposed relapses may receive limited benefit from these medications. New insights into the pathophysiology of progressive MS may lead us to new treatments through multiple alternative pathophysiologic pathways. Some early studies using this strategy show promise in slowing the progressive phase. Medication development for progressive MS faces multiple challenges due to lack of a single animal model demonstrating both pathology and clinical effects, absence of phase 1 surrogate biomarkers, and later phase trial endpoints that require large sample sizes and extended study durations. Nevertheless, the increase in number of trials and diversity of therapeutic approaches for progressive MS provides hope for effective therapy. Currently, the heterogeneity of the population with progressive MS requires an individualized treatment approach, and in some of these patients, stopping therapy may be a reasonable consideration. Symptomatic management remains critical for all patients with progressive MS as well as non-pharmacologic approaches that maximize quality of life.
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system, with recent estimates of around 1 million people living with MS in the US.1 In many countries, MS is a leading cause of disability among young adults, second only to trauma.2 Clinically, neurologic worsening (ie, disability) in MS can occur in the relapsing-remitting (RRMS) phase of disease due to incomplete recovery from neuroinflammatory relapses. However, in the 15% of patients with a progressive course from onset (PPMS), and in those with RRMS who transition to a secondary progressive phenotype (SPMS), neurologic worsening follows a slowly progressive pattern.3 A progressive disease course—either PPMS at onset or transitioning to SPMS—is the dominant factor affecting MS-related neurologic disability accumulation. In particular, epidemiologic studies have shown that, in the absence of transitioning to a progressive disease course, < 5% of individuals with MS will accumulate sufficient disability to necessitate use of a cane for ambulation.4-6 Therefore, developing disease modifying therapies (DMTs) that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS represent a critical unmet need.
Research into the development of DMTs for progressive MS has been hindered by a number of factors. In particular, the clinical definition and diagnosis of progressive MS has been an evolving concept. Diagnostic criteria for MS, which help facilitate the enrollment of appropriate subjects into clinical trials, have only recently clarified the current consensus definition for progressive MS—steadily increasing neurologic disability independent of clinical relapses. Looking back to the Schumacher criteria in 1965 and Poser criteria in 1983, it was acknowledged that neurologic symptoms in MS may follow a relapsing-remitting or progressive pattern, but little attempt was made to define progressive MS.7,8 The original McDonald criteria in 2001 defined diagnostic criteria for progressive MS.9 These criteria continued to evolve through subsequent revisions (eg, cerebrospinal fluid [CSF] specific oligoclonal bands no longer are an absolute requirement), and only in the 2017 revision was it emphasized that disability progression must occur independent of clinical relapses, concordant with similar emphasis in the 2013 revision of MS clinical course definitions.3,10
The interpretation of prior clinical trials of DMT for progressive MS must consider this evolving clinical definition. The US Food and Drug Administration (FDA) approved mitoxantrone in 2000—making it the first DMT to carry an approved label for SPMS. While achieving significant clinical efficacy, it is clear from the details of the trial that the enrolled subjects had a high degree of inflammatory disease activity, which suggests that mitoxantrone treats neuroinflammation and not relapse-independent worsening. More recently, disparate results were seen in the anti-CD20 (rituximab, ocrelizumab) and S1P receptor modulator (fingolimod, siponimod) trials targeted at patients with primary and secondary progressive MS.11-14 Although there are differences between these therapies, they are more similar than not within the same therapeutic class. Taken together, these trials illustrate the critical impact the narrower inclusion/exclusion criteria (namely age and extent of inflammatory activity) had on attaining positive outcomes. Other considerations, such as confounding illness, also may impact trial recruitment and generalizability of findings.
The lack of known biological targets in progressive MS, which is a complex disease with an incompletely understood and heterogeneous pathology, also hinders DMT development. Decades of research has characterized multifocal central nervous system (CNS) lesions that exhibit features of demyelination, inflammation, reactive gliosis, axonal loss, and neuronal damage. Until recently, however, much of this research focused on the relapsing phase of disease, and so the understanding of the pathologic underpinnings of progressive disease has remained limited. Current areas of investigation encompass a broad range of pathological processes, such as microglial activation, meningeal lymphoid follicles, remyelination failure, vulnerability of chronically demyelinated axons, oxidative injury, iron accumulation, mitochondrial damage, and others. There is the added complication that the pathologic processes underlying progressive MS are superimposed on the CNS aging process. In particular, the transition to progressive MS and the rate of disability accumulation during progressive MS show strong correlation with age.6,15-17
Finally, DMT development for progressive MS also is hindered by the lack of specific surrogate and clinical outcome measures. Trials for relapsing MS have benefited greatly from the relatively straightforward assessment of clinical relapses and inflammatory disease activity on magnetic resonance imaging (MRI). With the goal of developing DMTs that are highly effective at slowing or stopping the gradual accumulation of neurologic disability in progressive MS, which by definition occurs independent of clinical relapses, these measures are not directly relevant. The longitudinal clinical disability outcome measures change at a slower rate than in early, relapsing disease. The use of standardized scales (eg, the Expanded Disability Status Scale [EDSS]), lower limb function, upper limb function, cognition, or a combination is a subject of ongoing debate. For example, the ASCEND and IMPACT trials (placebo-controlled trials for SPMS with natalizumab and interferon β-1a, respectively) showed no significant impact in EDSS progression—but in both of these trials, the 9-hole peg test (9-HPT), a performance measure for upper limb function, showed significant improvement.10,18 Particularly in those with an EDSS of > 6.5, who are unlikely to have measurable EDSS progression, functional tests such as the 9-HPT or timed 25-foot walk may be more sensitive as measures for disability progression.11 MRI measures of brain atrophy is the current gold standard surrogate outcome for clinical trials in progressive MS, but others that may warrant consideration include optical coherence tomography (OCT) or CSF markers of axonal degeneration.
DMT for Progressive MS
Current diagnostic nomenclature separates patients with active (superimposed clinical and/or radiographic relapses) from those with inactive disease.3,12 Relapsing forms of MS include all RRMS and those with SPMS with superimposed relapses (ie, active SPMS). Following this paradigm shift, the FDA changed the indication for already approved DMT from RRMS to relapsing forms of MS. Below is a discussion of DMT that specifically use the term SPMS and PPMS in the indication, where phase 3 trial data for progressive MS is available.
In 2019, the FDA approved the first oral medication (siponimod) for active SPMS. Subsequently, updates to the labels of the older DMT expanded to include active SPMS. Until 2019, the only FDA approved medication for SPMS was mitoxantrone, and use of this medication was limited due to unfavorable adverse effects (AEs). No medications had obtained FDA approval for inactive SPMS to this point, which represented an unmet need for a considerable number of patients.
Mitoxantrone became the first DMT approved for use in SPMS in 2000 following early trials that showed reductions in EDSS worsening, change in ambulation index, reduced number of treated relapses, and prolonged time to first treated relapse. However, as with some of the other positive trials in progressive MS, it is difficult to discern the impact of suppression of relapses as opposed to direct impact on progressive pathophysiology. Within the placebo arm, for example, there were 129 relapses among the 64 subjects, which suggests that these cases had particularly active disease or were in the early stages of SPMS.13 Furthermore, the use of this medication was limited due to concerns of cardiotoxicity and hematologic malignancy as serious AEs.
The trials of interferon β-1b illustrate the same difficulty of isolating possible benefits in disease progression from disability accumulated from relapses. The first interferon β-1b trial for SPMS, was conducted in Europe using fingolimod and showed a delay in confirmed disability progression compared to placebo as measured with the EDSS.14 However, a North American trial that followed in 2004 was unable to replicate this finding.15 The patients in the European trial appeared to be in an earlier phase of SPMS with more active disease, and a post-hoc pooled analysis suggested that patients with active disease and those with more pronounced disability progression were more likely to benefit from treatment.16 Overall, interferons do not appear to appreciably alter disability in the long-term for patients with SPMS, though they may modify short-term, relapse-related disability.
Perhaps the most encouraging data for SPMS was found in the EXPAND trial, which investigated siponimod, an S1P receptor modulator that is more selective than fingolimod. The trial identified a 21% reduction in 3-month confirmed disability progression for SPMS patients taking siponimod compared with those taking a placebo.17 Although the patients in EXPAND did seem to have relatively less disease activity at baseline than those who participated in other SPMS trials, those who benefitted from siponimod were primarily patients who had clinical and/or radiographic relapses over the prior 2 years. Based on this, the FDA approved siponimod for active SPMS. The extent to which siponimod exerts a true neuroprotective effect beyond reducing inflammation has not been clearly established.
B-cell depleting therapies rituximab and ocrelizumab have been evaluated in both primary and secondary progressive MS populations. Early investigations of the chimeric rituximab in PPMS did not show benefits on disability (EDSS) progression; however, benefits were seen in analysis of some subgroups.18 With this in mind, the ORATORIO trial for the humanized version, ocreluzimab, included PPMS patients that were younger (aged < 55 years) and had cutoffs for disease duration (< 15 years for those with EDSS more than 5 years, < 10 years for those with EDSS less than 5 years). The study showed statistically significant changes on disability progression, which led to ocrelizumab receiving the first FDA indication for PPMS.11 There are substantial pathophysiologic similarities between PPMS and SPMS in the progressive phase.19 While these medications may have similar effects across these disease processes, these benefits have not yet been demonstrated in a prospective trial for the SPMS population. Regardless, B-cell depleting therapy is a reasonable consideration for select patients with active SPMS, consistent with a relapsing form of MS.
Therapies in Development
DMT development for progressive MS is a high priority area. Current immunomodulatory therapies for RRMS have consistently been ineffective in the inactive forms of MS, with the possible exceptions of ocrelizumab and siponimod. Therefore, instead of immunosuppression, many agents currently in phase 2 and 3 clinical trials target alternative pathophysiological processes in order to provide neuroprotection, and/or promote remyelination and neurogenesis. These targets include oxidative stress (OS), non-T cell mediated inflammation, and mitochondrial/energy failure.20 Below we review a selection of clinical trials testing agents following these approaches. Many agents have more than one potential mechanism of action (MOA) that could benefit progressive MS.
Lipoic acid (LA), also known as α-lipoic acid and thiotic acid, is one such agent targeting alternative pathophysiology in SPMS. LA is an endogenous agent synthesized de novo from fatty acids and cysteine as well as obtained in small amounts from foods.21 It has antioxidant (AO) properties including direct radical scavenging, regeneration of glutathione, and upregulation of AO enzymes via the NrF2 pathway.22 It supports mitochondria as a key cofactor for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, and it also aids mitochondrial DNA synthesis.21,22 Studies in experimental autoimmune encephalomyelitis, a widely used experimental mouse model of inflammatory demyelinating disease, also indicate a reduction in excessive microglial activation.23 A phase 2 pilot randomized controlled trial (RCT) of 1200 mg LA in SPMS (n = 51) resulted in significantly less whole brain atrophy by SIENA (Structural Image Evaluation, Using Normalization, of Atrophy) at 2 years.24 A follow-up multicenter trial is ongoing.
Simvastatin also targets alternative pathophysiology in SPMS. It has anti-inflammatory effects, improves vascular function, and promotes neuroprotection by reducing excitotoxicity. A phase 2 RCT demonstrated a reduction in whole brain atrophy in SPMS (n = 140), and a phase 3 trial is underway.25 Ibudilast is another repurposed drug that targets alternative inflammation by inhibiting several cyclic nucleotide phosphodiesterases, macrophage migration inhibitory factor and toll-like receptor 4. A phase 2 trial (n = 225) in both SPMS and PPMS also demonstrated a reduction in brain atrophy, but participants had high rates of AEs.26
Lithium and riluzole promote neuroprotection by reducing excitotoxicity. Lithium is a pharmacologic active cation used as a mood stabilizer and causes inhibition of glycogen synthase kinase-3β. Animal models also indicate that lithium may decrease inflammation and positively impact neurogenesis.27 A crossover pilot trial demonstrated tolerability with trends toward stabilization of EDSS and reductions in brain atrophy.28 Three neuroprotective agents, riluzole (reduces glutamate excitotoxicity), fluoxetine (stimulates glycogenolysis and improves mitochondrial energy production), and amiloride (an acid-sensing ion channel blocker that opens in response to inflammation) were tested in a phase 2b multi-arm, multi-site parallel group RCT in SPMS (n = 445). The study failed to yield differences from placebo for any agent in reduction of brain volume loss.29 A prior study of lamotrigine, a sodium channel blocker, also failed to find changes in brain volume loss.30 These studies highlight the large sample sizes and/or long study durations needed to test agents using brain atrophy as primary outcome. In the future, precise surrogate markers of neuroprotection will be a great need for earlier phase trials. These results also suggest that targeting > 1 MOA may be necessary to treat SPMS effectively.
Efforts to promote remyelination target one hallmark of MS damage. High dose biotin (about 10,000× usual dose) may promote myelin repair as a cofactor for fatty acid synthesis and support mitochondrial oxidative phosphorylation. While a RCT yielded a greater proportion of participants with either PPMS or SPMS with improvement in disability than placebo at 12 months, an open label trial suggested otherwise indicating a need for a more definitive trial.31,32
Anti-LINGO-1 (opicinumab) is a monoclonal antibody that targets LINGO, a potent negative regulator of oligodendrocyte differentiation and myelination.33 Although this agent failed in a phase 2 trial in relapsing MS, and is thus unlikely to be tested in progressive forms, the innovative approach to stimulating oligodendrocytes is ongoing. One such effort is to use thyroid hormone, crucial to myelin formation during development, as a repair agent in MS.34 A dose-finding study of thyroid hormone was completed and efforts to develop a thyromimetic agent are ongoing.
Finally, efforts to promote neurogenesis remain a goal for many neurodegenerative diseases. Exercise appears to prevent age-related atrophy of the hippocampus in animals and humans and help maintain neuronal health.35 In patients with RRMS, cortical thickness is impacted positively by resistance training, which suggests a neuroprotective effect.36 A multi-center trial of exercise in patients with progressive MS investigating cognitive outcomes is ongoing.
Discontinuing DMT
In the early 1990s, the successful development of immune modulating therapies that reliably reduced disease activity in RRMS led to widespread initiation in patients with relapsing disease. However, guidance on when or if to discontinue DMT, even in those who have transitioned to SPMS, remains largely absent at this time. Requests to discontinue DMT may come from patients weary of taking medication (especially injections), bothered by AEs, or those who no longer perceive efficacy from their treatments. Clinicians also may question the benefit of immune modulation in patients with longstanding freedom from relapses or changes in MRI lesion burden.
To inform discussion centered on treatment discontinuation, a clinical trial is currently underway to better answer the question of when and how to withdraw MS therapy. Discontinuation of Disease Modifying Therapies in Multiple Sclerosis (DISCO-MS) is a prospective, placebo-controlled RCT and its primary endpoint is recurrence of disease activity over a 2 year follow-up period.37 Eligibility requirements for the trial include age > 55 years, 5-year freedom from relapses, and 3-year freedom from new MRI lesions (criteria informed by progressive MS cohort studies).31 In addition to demonstrating the active disease recurrence rates in this patient population, the trial also aims to identify risk factors for recurrent disease activity among treated MS patients.37 DISCO-MS builds upon a series of retrospective and observational studies that partially answered these questions, albeit in the context of biases inherent in retrospective or observational studies.
A Minneapolis MS Treatment and Research Center single-center study identified 77 SPMS patients with no acute CNS inflammatory events over 2 to 20 years and advised these patients to stop taking DMT.32 In this group, 11.7% of subjects experienced recurrent active disease. Age was the primary discriminating factor. The mean age of those who experienced disease activity was 56 years vs 61 years those who did not. A second observational study from France found that among 100 SPMS patients treated either with interferon β or glatiramer acetate for at least 6 months, 35% experienced some form of inflammatory disease upon discontinuation.38 Sixteen patients relapsed and 19 developed gadolinium-enhancing MR lesions after DMT discontinuation. However, the age of the cohort was younger than the Minneapolis study (47.2 years vs 61 years), and reasons for discontinuation (eg, AEs or lack of disease activity) were not considered in the analysis.
Other studies examining the safety of DMT discontinuation have not considered MS subtype or excluded patients with progressive subtypes of MS. The largest studies to date on DMT discontinuation utilized the international MSBase global patient registry, which identified nearly 5,000 patients who discontinued interferons (73%), glatiramer acetate (18%), natalizumab (6%), or fingolimod (3%), without specifying the reasons for discontinuation.39 Despite these shortcomings, data reveal trends that are helpful in predicting how MS tends to behave in patients who have discontinued therapy. Not surprisingly, disability progression was most likely among patients already characterized as having a progressive phenotype, while relapses were less likely to occur among older, progressive patients.
Although clinicians may be increasingly willing to discuss DMT discontinuation with their patients, at least 1 study exploring patient perspectives on stopping treatment suggests widespread reluctance to stop treatment. A survey conducted with participants in the North American Research Committee on Multiple Sclerosis patient-report registry found that among survey respondents, only 11.9% would discontinue their MS medication if deemed stable, while 66.3% stated they were unlikely to stop treatment.40
These results suggest that before clinicians incorporate DMT discontinuation into the normal course of discussion with patients, they should be prepared to provide both education (on the wisdom of stopping under the right circumstances) and evidence to support when and how to make the recommendation. Based on existing evidence, criteria for recommending treatment discontinuation might include prolonged freedom from disease activity (≥ 5 years), age > 55 years or 60 years, and a progressive disease course. Thus far, no combination of factors has been shown to completely predict an event-free transition off of medicine. Since no fixed algorithm yet exists to guide DMT stoppage in MS, reasonable suggestions for monitoring patients might include surveillance MRIs, more frequent clinic visits, and possible transitional treatment for patients coming off of natalizumab or fingolimod, since these drugs have been associated with rebound disease activity when discontinued.41,42
Clinicians wishing to maximize function and quality of life for their patients at any age or stage of disease should look to nonpharmacologic interventions to lessen disability and maximize quality of life. While beyond the scope of this discussion, preliminary evidence suggests multimodal (aerobic, resistance, balance) exercise may enhance endurance and cognitive processing speed, and that treatment of comorbid diseases affecting vascular health benefits MS. 43
Conclusions
The development of numerous treatments for RRMS has established an entirely new landscape and disease course for those with MS. While this benefit has not entirely extended to those with progressive MS, those with active disease with superimposed relapses may receive limited benefit from these medications. New insights into the pathophysiology of progressive MS may lead us to new treatments through multiple alternative pathophysiologic pathways. Some early studies using this strategy show promise in slowing the progressive phase. Medication development for progressive MS faces multiple challenges due to lack of a single animal model demonstrating both pathology and clinical effects, absence of phase 1 surrogate biomarkers, and later phase trial endpoints that require large sample sizes and extended study durations. Nevertheless, the increase in number of trials and diversity of therapeutic approaches for progressive MS provides hope for effective therapy. Currently, the heterogeneity of the population with progressive MS requires an individualized treatment approach, and in some of these patients, stopping therapy may be a reasonable consideration. Symptomatic management remains critical for all patients with progressive MS as well as non-pharmacologic approaches that maximize quality of life.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data [published correction appears in Neurology. 2019;93(15):688]. Neurology. 2019;92(10):e1029-e1040.
2. Browne P, Chandraratna D, Angood C, et al. Atlas of multiple sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83(11):1022-1024.
3. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
4. Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112(Pt 1):133-146. 5. Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain. 2006;129(Pt 3):595-605.
6. Tutuncu M, Tang J, Zeid NA, et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult Scler. 2013;19(2):188-198.
7. Schumacher GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci. 1965;122:552-568.
8. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13(3):227-231.
9. McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121-127.
10. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
11. Montalban X, Hauser SL, Kappos L, et al; ORATORIO Clinical Investigators. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209-220.
12. Hawker K, O’Connor P, Freedman MS, et al; OLYMPUS trial group. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
13. Kappos L, Bar-Or A, Cree BAC, et al; EXPAND Clinical Investigators. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study [published correction appears in Lancet. 2018;392(10160):2170]. Lancet. 2018;391(10127):1263-1273.
14. Lublin F, Miller DH, Freedman MS, et al; INFORMS study investigators. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial [published correction appears in Lancet. 2017;389(10066):254]. Lancet. 2016;387(10023):1075-1084.
15. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343(20):1430-1438.
16. Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(Pt 3):584-594.
17. Leray E, Yaouanq J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(Pt 7):1900–1913.
18. Kapoor R, Ho PR, Campbell N, et al; ASCEND investigators. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405-415.
19. Koch MW, Mostert J, Uitdehaag B, Cutter G. Clinical outcome measures in SPMS trials: an analysis of the IMPACT and ASCEND original trial data sets [published online ahead of print, 2019 Sep 13]. Mult Scler. 2019;1352458519876701.
20. Hartung HP, Gonsette R, König N, et al; Mitoxantrone in Multiple Sclerosis Study Group (MIMS). Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018-2025.
21. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352(9139):1491-1497.
22. Gorąca A, Huk-Kolega H, Piechota A, Kleniewska P, Ciejka E, Skibska B. Lipoic acid - biological activity and therapeutic potential. Pharmacol Rep. 2011;63:849-858.
23. Chaudhary P, Marracci G, Pocius E, Galipeau D, Morris B, Bourdette D. Effects of lipoic acid on primary murine microglial cells. J Neuroimmunol. 2019;334:576972.
24. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4:e374.
25. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213-2221.
26. Fox RJ, Coffey CS, Conwit R, et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med. 2018;379:846-855.
27. Rinker JR, 2nd, Cossey TC, Cutter GR, Culpepper WJ. A retrospective review of lithium usage in veterans with multiple sclerosis. Mult Scler Relat Disord. 2013;2:327-333.
28. Rinker JR, W Meador, V Sung, A Nicholas, G Cutter. Results of a pilot trial of lithium in progressive multiple sclerosis. ECTRIMS Online Library. 09/16/16; 145965; P12822016.
29. Chataway J, De Angelis F, Connick P, et al; MS-SMART Investigators. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): a phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020;19(3):214-225.
30. Kapoor R, Furby J, Hayton T, et al. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9:681-688.
31. Paz Soldan MM, Novotna M, Abou Zeid N, et al. Relapses and disability accumulation in progressive multiple sclerosis. Neurology. 2015;84:81-88.
32. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19:11-14.
33. Ruggieri S, Tortorella C, Gasperini C. Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev Neurother 2017;17:1081-1089.
34. Hartley MD, Banerji T, Tagge IJ, et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. 2019;4(8):e126329.
35. Firth J, Stubbs B, Vancampfort D, et al. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage. 2018;166:230-238.
36. Kjolhede T, Siemonsen S, Wenzel D, et al. Can resistance training impact MRI outcomes in relapsing-remitting multiple sclerosis? Mult Scler. 2018;24:1356-1365.
37. US National Library of Medicine, Clinicaltrials.gov. Discontinuation of Disease Modifying Therapies (DMTs) in Multiple Sclerosis (MS) (DISCOMS). https://clinicaltrials.gov/ct2/show/NCT03073603. Updated February 10, 2020. Accessed March 26, 2020.
38. Bonenfant J, Bajeux E, Deburghgraeve V, Le Page E, Edan G, Kerbrat A. Can we stop immunomodulatory treatments in secondary progressive multiple sclerosis? Eur J Neurol. 2017;24:237-244.
39. Kister I, Spelman T, Patti F, et al. Predictors of relapse and disability progression in MS patients who discontinue disease-modifying therapy. J Neurol Sci. 2018;391:72-76.
40. McGinley MP, Cola PA, Fox RJ, Cohen JA, Corboy JJ, Miller D. Perspectives of individuals with multiple sclerosis on discontinuation of disease-modifying therapies. Mult Scler. 2019:1352458519867314.
41. Hatcher SE, Waubant E, Graves JS. Rebound Syndrome in Multiple Sclerosis After Fingolimod Cessation-Reply. JAMA Neurol. 2016;73:1376.
42. Vellinga MM, Castelijns JA, Barkhof F, Uitdehaag BM, Polman CH. Postwithdrawal rebound increase in T2 lesional activity in natalizumab-treated MS patients. Neurology. 2008;70:1150-1151.
43. Sandroff BM, Bollaert RE, Pilutti LA, et al. Multimodal exercise training in multiple sclerosis: A randomized controlled trial in persons with substantial mobility disability. Contemp Clin Trials 2017;61:39-47.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data [published correction appears in Neurology. 2019;93(15):688]. Neurology. 2019;92(10):e1029-e1040.
2. Browne P, Chandraratna D, Angood C, et al. Atlas of multiple sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83(11):1022-1024.
3. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
4. Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112(Pt 1):133-146. 5. Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain. 2006;129(Pt 3):595-605.
6. Tutuncu M, Tang J, Zeid NA, et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult Scler. 2013;19(2):188-198.
7. Schumacher GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci. 1965;122:552-568.
8. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13(3):227-231.
9. McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121-127.
10. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
11. Montalban X, Hauser SL, Kappos L, et al; ORATORIO Clinical Investigators. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209-220.
12. Hawker K, O’Connor P, Freedman MS, et al; OLYMPUS trial group. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
13. Kappos L, Bar-Or A, Cree BAC, et al; EXPAND Clinical Investigators. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study [published correction appears in Lancet. 2018;392(10160):2170]. Lancet. 2018;391(10127):1263-1273.
14. Lublin F, Miller DH, Freedman MS, et al; INFORMS study investigators. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial [published correction appears in Lancet. 2017;389(10066):254]. Lancet. 2016;387(10023):1075-1084.
15. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343(20):1430-1438.
16. Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(Pt 3):584-594.
17. Leray E, Yaouanq J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(Pt 7):1900–1913.
18. Kapoor R, Ho PR, Campbell N, et al; ASCEND investigators. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405-415.
19. Koch MW, Mostert J, Uitdehaag B, Cutter G. Clinical outcome measures in SPMS trials: an analysis of the IMPACT and ASCEND original trial data sets [published online ahead of print, 2019 Sep 13]. Mult Scler. 2019;1352458519876701.
20. Hartung HP, Gonsette R, König N, et al; Mitoxantrone in Multiple Sclerosis Study Group (MIMS). Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018-2025.
21. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352(9139):1491-1497.
22. Gorąca A, Huk-Kolega H, Piechota A, Kleniewska P, Ciejka E, Skibska B. Lipoic acid - biological activity and therapeutic potential. Pharmacol Rep. 2011;63:849-858.
23. Chaudhary P, Marracci G, Pocius E, Galipeau D, Morris B, Bourdette D. Effects of lipoic acid on primary murine microglial cells. J Neuroimmunol. 2019;334:576972.
24. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4:e374.
25. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213-2221.
26. Fox RJ, Coffey CS, Conwit R, et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med. 2018;379:846-855.
27. Rinker JR, 2nd, Cossey TC, Cutter GR, Culpepper WJ. A retrospective review of lithium usage in veterans with multiple sclerosis. Mult Scler Relat Disord. 2013;2:327-333.
28. Rinker JR, W Meador, V Sung, A Nicholas, G Cutter. Results of a pilot trial of lithium in progressive multiple sclerosis. ECTRIMS Online Library. 09/16/16; 145965; P12822016.
29. Chataway J, De Angelis F, Connick P, et al; MS-SMART Investigators. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): a phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020;19(3):214-225.
30. Kapoor R, Furby J, Hayton T, et al. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9:681-688.
31. Paz Soldan MM, Novotna M, Abou Zeid N, et al. Relapses and disability accumulation in progressive multiple sclerosis. Neurology. 2015;84:81-88.
32. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19:11-14.
33. Ruggieri S, Tortorella C, Gasperini C. Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev Neurother 2017;17:1081-1089.
34. Hartley MD, Banerji T, Tagge IJ, et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. 2019;4(8):e126329.
35. Firth J, Stubbs B, Vancampfort D, et al. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage. 2018;166:230-238.
36. Kjolhede T, Siemonsen S, Wenzel D, et al. Can resistance training impact MRI outcomes in relapsing-remitting multiple sclerosis? Mult Scler. 2018;24:1356-1365.
37. US National Library of Medicine, Clinicaltrials.gov. Discontinuation of Disease Modifying Therapies (DMTs) in Multiple Sclerosis (MS) (DISCOMS). https://clinicaltrials.gov/ct2/show/NCT03073603. Updated February 10, 2020. Accessed March 26, 2020.
38. Bonenfant J, Bajeux E, Deburghgraeve V, Le Page E, Edan G, Kerbrat A. Can we stop immunomodulatory treatments in secondary progressive multiple sclerosis? Eur J Neurol. 2017;24:237-244.
39. Kister I, Spelman T, Patti F, et al. Predictors of relapse and disability progression in MS patients who discontinue disease-modifying therapy. J Neurol Sci. 2018;391:72-76.
40. McGinley MP, Cola PA, Fox RJ, Cohen JA, Corboy JJ, Miller D. Perspectives of individuals with multiple sclerosis on discontinuation of disease-modifying therapies. Mult Scler. 2019:1352458519867314.
41. Hatcher SE, Waubant E, Graves JS. Rebound Syndrome in Multiple Sclerosis After Fingolimod Cessation-Reply. JAMA Neurol. 2016;73:1376.
42. Vellinga MM, Castelijns JA, Barkhof F, Uitdehaag BM, Polman CH. Postwithdrawal rebound increase in T2 lesional activity in natalizumab-treated MS patients. Neurology. 2008;70:1150-1151.
43. Sandroff BM, Bollaert RE, Pilutti LA, et al. Multimodal exercise training in multiple sclerosis: A randomized controlled trial in persons with substantial mobility disability. Contemp Clin Trials 2017;61:39-47.
Multiple Sclerosis Medications in the VHA: Delivering Specialty, High-Cost, Pharmacy Care in a National System (FULL)
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
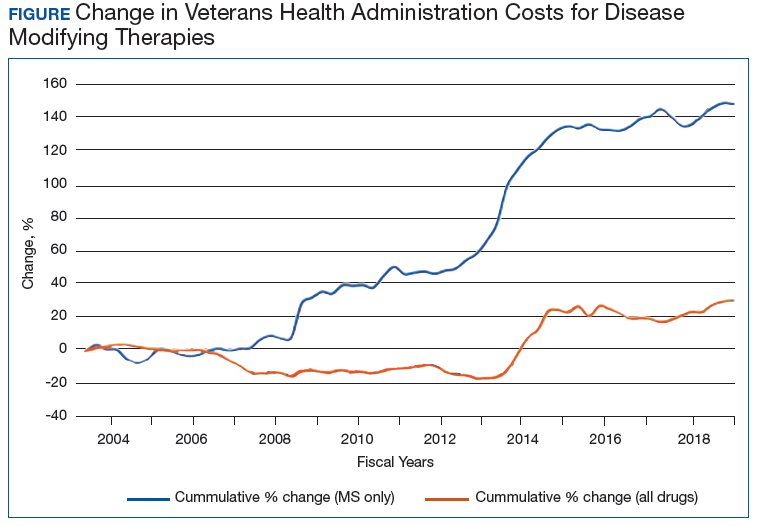
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
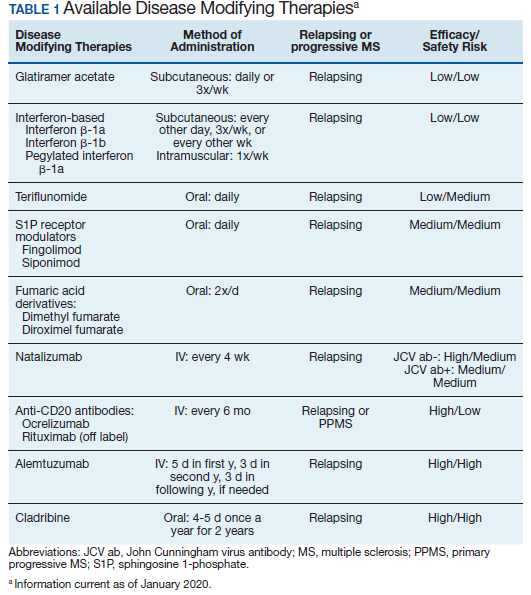
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
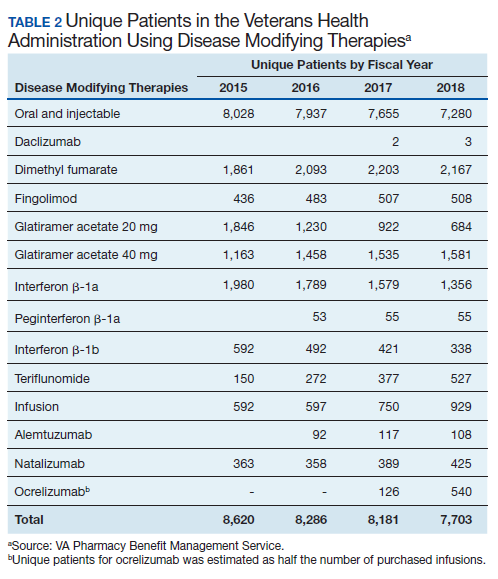
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
Immunotherapies Targeting α -Synuclein in Parkinson Disease
Parkinson disease (PD) is a progressive neurodegenerative disorder, characterized by diverse clinical symptoms. PD can present with rest tremor, bradykinesia, rigidity, falls, postural instability, and multiple nonmotor symptoms. Marras and colleagues estimated in a comprehensive meta-analysis that there were 680,000 individuals with PD in the US in 2010; this number is expected to double by 2030 based on the US Census Bureau population projections.1 An estimated 110,000 veterans may be affected by PD; hence, understanding of PD pathology, clinical progression, and effective treatment strategies is of paramount importance to the Veterans Health Administration (VHA).2
The exact pathogenesis underlying clinical features is still being studied. Pathologic diagnosis of PD relies on loss of dopamine neurons in the substantia nigra and accumulation of the abnormal protein, α-synuclein, in the form of Lewy bodies and Lewy neurites. Lewy bodies and neurites accumulate predominantly in the substantia nigra in addition to other brain stem nuclei and cerebral cortex. Lewy bodies are intraneuronal inclusions with a hyaline core and a pale peripheral halo. Central core stains positive for α-synuclein.3,4 Lewy neurites are widespread and are believed to play a larger role in the pathogenesis of PD compared with those of Lewy bodies.5
α-Synuclein
α-synuclein is a small 140 amino-acid protein with a N-terminal region that can interact with cell membranes and a highly acidic unstructured C-terminal region.6 α-synuclein is physiologically present in the presynaptic terminals of neurons and involved in synaptic plasticity and vesicle trafficking.7 There are different hypotheses about the native structure of α-synuclein. The first suggests that it exists in tetrameric form and may be broken down to monomer, which is the pathogenic form of α-synuclein. The second hypothesis suggests that it exists primarily in monomeric form, whereas other studies have shown that both forms exist and with pathologic changes, monomer accumulates in abundance and is neurotoxic.8-11 Work by Burré and colleagues shows that native α-synuclein exists in 2 forms: a soluble, cytosolic α-synuclein, which is monomeric, and a membrane-bound multimeric form.12,13
Alteration in aggregation properties of this protein is believed to play a central role in the pathogenesis of PD.14,15 Pathologic α-synuclein exists in insoluble forms that can aggregate into oligomers and fibrillar structures.16 Lysosomal dysfunction may promote accumulation of insoluble α-synuclein. Prior work has shown that several degradation pathways in lysosomes, including the ubiquitin-proteasome system and autophagy-lysosomal pathway, are down regulated, thus contributing to the accumulation of abnormal α-synuclein.17,18 Accumulation of pathologic α-synuclein leads to mitochondrial dysfunction in PD animal models, contributing further to neurotoxicity.19,20 Aggregates of phosphorylated α-synuclein have been demonstrated in dementia with Lewy body.21
In addition, α-synuclein aggregates may be released into extracellular spaces to be taken up by adjacent cells, where they can cause further misfolding and aggregation of protein.22 Previous work in animal models suggested a prion proteinlike spread of α-synuclein.23 This finding can have long-term therapeutic implications, as preventing extracellular release of abnormal form of α-synuclein will prevent the spread of pathologic protein. This can form the basis of neuroprotection in patients with PD.24
It has been proposed that α-synuclein accumulation and extracellular release initiates an immune response that leads to activation of microglia. This has been shown in PD animal models, overexpressing α-synuclein. In 2008 Park and colleagues demonstrated that microglial activation is enhanced by monomeric α-synuclein, not by the aggregated variant.25 Other studies have reported activated microglia around dopaminergic cells in substantia nigra.26 Sulzer and colleagues showed that peptides from α-synuclein can act as antigens and trigger an autoimmune reaction via T cells.27 PD may be associated with certain HLA-haplotypes.28 In other words, α-synuclein can induce neurodegeneration via different mechanisms, including alteration in synaptic vesicle transmission, mitochondrial dysfunction, neuroinflammation, and induction of humoral immunity.
Immunization
Due to these observations, there had been huge interest in developing antibody-based therapies for PD. A similar approach had been tested in Alzheimer disease (AD). Intracellular tangles of tau protein and extracellular aggregates of amyloid are the pathologic substrates in AD. Clinical trials utilizing antibodies targeting amyloid showed reduction in abnormal protein accumulation but no significant improvement in cognition.29 In addition, adverse events (AEs), such as vasogenic edema and intracerebral hemorrhage, were reported.30 Careful analysis of the data suggested that inadequate patient selection or targeting only amyloid, may have contributed to unfavorable results.31 Since then, more recent clinical trials have focused on careful patient selection, use of second generation anti-amyloid antibodies and immunotherapies targeting tau.32
Several studies have tested immunotherapies in PD animal models with the aim of targeting α-synuclein. Immunotherapies can be instituted in 2 ways: active immunization in which the immune system is stimulated to produce antibodies against α-synuclein or passive immunization in which antibodies against α-synuclein are administered directly. Once α-synuclein antibodies have crossed the blood-brain barrier, they are hypothesized to clear the existing α-synuclein. Animal studies have demonstrated the presence of these antibodies within the neurons. The mechanism of entry is unknown. Once inside the cells, the antibodies activate the lysosomal clearance, affecting intracellular accumulation of α-synuclein. Extracellularly, they can bind to receptors on scavenger cells, mainly microglia, activating them to facilitate uptake of extracellular α-synuclein. Binding of the antibodies to α-synuclein directly prevents the uptake of toxic protein by the cells, blocking the transfer and spread of PD pathology.33
Active Immunization
Active immunization against α-synuclein was demonstrated by Masliah and colleagues almost a decade ago. They administered recombinant human α-synuclein in transgenic mice expressing α-synuclein under the control of platelet-derived growth factor β. Reduction of accumulated α-synuclein in neurons with mild microglia activation was noted. It was proposed that the antibodies produced were able to bind to abnormal α-synuclein, were recognized by the lysosomal pathways, and degraded.34 Ghochikyan and colleagues developed vaccines by using α-synuclein-derived peptides. This induced formation of antibodies against α-synuclein in Lewy-bodies and neurites.35 Over time, other animal studies have been able to expand on these results.36
AFFiRiS, an Austrian biotechnology company, has developed 2 peptide vaccines PD01A and PD03A. Both peptides when administered to PD animal models caused antibody-based immune response against aggregated α-synuclein. Humoral autoimmune response was not observed in these studies; no neuroinflammation or neurotoxicity was noted. These peptides did not affect levels of physiologic α-synuclein, targeting only the aggregated form.37 These animal models showed improved motor and cognitive function. Similar results were noted in multiple system atrophy (MSA) animal models.38,39
The first human phase 1, randomized, parallel-group, single-center study recruited 32 subjects with early PD. Twelve subjects each were included in low- or high-dose treatment group, and 8 were included in the control group. Test subjects randomly received 4 vaccinations of low- or high-dose PD01A. Both doses were well tolerated, and no drug-related serious AEs were reported. The study confirmed the tolerability and safety of subcutaneous PD01A vaccine administration. These subjects were included in a 12-month, phase 1b follow-up extension study, AFF008E. In 2018, it was reported that administration of 6 doses of PD01A, 4 primary and 2 booster immunization, was safe. The vaccine showed a clear immune response against the peptide and cross-reactivity against α-synuclein targeted epitope. Booster doses stabilized the antibody titers. Significant increase in antibody titers against PD01A was seen over time, which was translated into a humoral immune response against α-synuclein. In addition, PD01A antibodies also were reported in cerebrospinal fluid.40
AFFiRiS presented results of a phase 1 randomized, placebo-controlled trial in 2017, confirming the safety of PD03A in patients with PD. The study showed a clear dose-dependent immune response against the peptide and cross-reactivity against α-synuclein targeted epitope.41 AFFiRiS recently presented results of another phase 1 clinical study assessing the safety and tolerability of vaccines PD01A and PD03A in patients with early MSA. Both vaccines were well tolerated, and PD01A induced an immune response against the peptide and α-synuclein epitope.42 These results have provided hope for further endeavors to develop active immunization strategies for PD.
Passive Immunization
Passive immunization against α-synuclein was first reported by Masliah and colleagues in 2011. A monoclonal antibody against the C-terminus of α-synuclein, 9E4, was injected into a transgenic mouse model of PD. There was reduction in α-synuclein aggregates in the brain along with improvement in motor and cognitive impairment.43 The C-terminus of α-synuclein plays a key role in the pathogenesis of PD. Changes in the C-terminus of α-synuclein induces formation of α-synuclein oligomers and subsequent neuronal spread. Antibody binds to the C-terminus and prevents structural changes that can lead to oligomerization of α-synuclein. Since the first study by Masliah, few other immunization studies utilized different antibodies against the C-terminus of α-synuclein. It was shown in a mouse model that binding of such antibodies promoted clearance of the α-synuclein by microglia.44
Based on these animal studies, Prothena Biosciences (South San Francisco, CA) designed a phase 1, double-blind, randomized, placebo-controlled clinical trial of prasinezumab (investigational monoclonal antibody against C-terminus of α-synuclein), in subjects without PD. The results showed that it was well tolerated, and there was dose-dependent reduction in the levels of free
BIIB054 is another monoclonal antibody that targets the N-terminal of α-synuclein. In animal models, antibodies targeting the N-terminus reduced α-synuclein triggered cell death and reduced the number of activated microglia.48 BIIB054, from Biogen (Cambridge, MA), was studied in 40 healthy subjects and was well tolerated with a favorable safety profile and could cross the blood-brain barrier. Like the prasinezumab study, this also was an ascending-dose study to assess safety and tolerability. In 2018, a randomized, double-blind, placebo-controlled, single-ascending dose study in patients with PD reported that BIIB054 was well tolerated, and the presence of BIIB054-synuclein complexes in the plasma were confirmed.49 A phase 2, multicenter, randomized, double-blind, placebo-controlled study (SPARK) with an active-treatment dose-blinded period, designed to evaluate the safety, pharmacokinetics, and the pharmacodynamics of BIIB054 is currently recruiting patients with PD.
Finally, BioArctic (Stockholm, Sweden) developed antibodies that are selective for oligomeric forms of α-synuclein, which it licensed to AbbVie (North Chicago, Il).50 These antibodies do not target the N- or C-terminus of α-synuclein. Since α-synuclein oligomers play an important role in the pathogenesis of PD, targeting them with antibodies at an early stage may prove to be an effective strategy for removal of pathogenic α-synuclein. Clinical trials are forthcoming.
Conclusions
Immunotherapy against α-synuclein has provided a new therapeutic avenue in the field of neuroprotection. Results from the first human clinical trial are promising, but despite these results, more work is needed to clarify the role of α-synuclein in the pathogenesis of PD in humans. Most of the work concerning α-synuclein aggregation and propagation has been reported in animal models. Whether similar process exists in humans is a debatable question. Similarly, more knowledge is needed about how and where in the human brain antibodies act to give neuroprotective effects. Timing of administration of immunotherapies in real time will be a crucial question.
PD is clinically evident once 80% of dopaminergic neurons in substantia nigra are lost due to neurodegeneration. Should immunotherapy be administered to symptomatic patients with PD, or if it will be beneficial only for presymptomatic, high-risk patients needs to be determined. Like AD trials, not only careful selection of patients, but determination of optimal timing for treatment will be essential. As the understanding of PD pathogenesis and therapeutics evolves, it will become clear whether immunization targeting α-synuclein will modify disease progression.
1. Marras C, Beck JC, Bower JH, et al; Parkinson’s Foundation P4 Group. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018;4(1):21. doi:10.1038/s41531-018-0058-0
2. Mantri S, Duda JE, Morley JF. Early and accurate identification of Parkinson disease among US veterans. Fed Pract. 2019;36(suppl 4):S18-S23. doi:10.12788/fp.37-0034
3. Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7(suppl 1):S71-S85. doi:10.3233/JPD-179001
4. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839-840. doi:10.1038/42166
5. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197-211. doi:10.1016/s0197-4580(02)00065-9
6. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79(6):1044-1066. doi:10.1016/j.neuron.2013.09.004
7. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663-1667. doi:10.1126/science.1195227
8. Binolfi A, Fernández CO, Sica MP, Delfino JM, Santos J. Recognition between a short unstructured peptide and a partially folded fragment leads to the thioredoxin fold sharing native-like dynamics. Proteins. 2012;80(5):1448-1464. doi:10.1002/prot.24043
9. Fauvet B, Mbefo MK, Fares MB, et al. α-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345-15364. doi:10.1074/jbc.M111.318949.
10. Wang W, Perovic I, Chittuluru J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108(43):17797-17802. doi:10.1073/pnas.1113260108
11. Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P. From α-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012;1476:183-202. doi:10.1016/j.brainres.2012.04.014
12. Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native α-synuclein. Nature. 2013;498(7453):E4-E7.
13. Burré J, Sharma M, Südhof TC. α-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA. 2014;111(40):E4274-E4283. doi:10.1073/pnas.1416598111
14. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1-13. doi:10.1038/nm.4269
15. Burré J, Sharma M, Südhof TC. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci. 2015;35(13):5221-5232. doi:10.1523/JNEUROSCI.4650-14.2015
16. Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24(8):1888-1896. doi:10.1523/JNEUROSCI.3809-03.2004
17. Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L . α-synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem. 2004;279(45):46915-46920. doi:10.1074/jbc.M405146200
18. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200-210. doi:10.1016/j.tibs.2015.02.003
19. Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochem Biophys Acta. 2010;1802(1):29-44. doi:10.1016/j.bbadis.2009.08.013
20. Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein: a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92-98. doi:10.1038/nrneurol.2013.275
21. Colom-Cadena M, Pegueroles J, Herrmann AG, et al. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain. 2017;140(12):3204-3214. doi:10.1093/brain/awx275
22. Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57-71. doi:10.1016/j.neuron.2011.08.033
23. Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(pt 4):1128-1138. doi:10.1093/brain/awt037
24. Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22-33. doi:10.1016/j.pharmthera.2016.11.010
25. Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56(11):1215-1223. doi:10.1002/glia.20691
26. Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimmune Pharmacol. 2013;8(1):189-201. doi:10.1007/s11481-013-9435-y
27. Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546(7660):656-661. doi:10.1038/nature22815
28. Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genetics. 2010;42(9):781-785. doi:10.1038/ng.642
29. Holmes C, Boche D, Wilkinson D, et al. Long term effects of Aβ42 immunisation in Alzheimer’s disease: follow up of a randomized, placebo-controlled phase I trial. Lancet. 2008;372(9634):216-223. doi:10.1016/S0140-6736(08)61075-2
30. Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241-249. doi:10.1016/S1474-4422(12)70015-7
31. Wisniewski T, Goñi F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499-507. doi:10.1016/j.bcp.2013.12.020
32. Herline K, Drummond E, Wisniewski T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines. 2018;17(8):707-721. doi:10.1080/14760584.2018.1500905
33. Bergstrom AL, Kallunki P, Fog K. Development of passive immunotherapies for synucleopathies. Mov Disord. 2015;31(2):203-213. doi:10.1002/mds.26481
34. Masliah E, Rockenstein E, Adame A, et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857-868. doi:10.1016/j.neuron.2005.05.010
35. Ghochikyan A, Petrushina I, Davtyan H, et al. Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: feasibility study. Neurosci Lett. 2014;560:86-91. doi:10.1016/j.neulet.2013.12.028
36. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. α-synuclein vaccination prevents the accumulation of Parkinson’s disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72(7):624-645. doi:10.1097/NEN.0b013e31829768d2
37. Mandler M, Valera E, Rockenstein E, et al. Next generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127(6):861-879. doi:10.1007/s00401-014-1256-4
38. Mandler M, Valera E, Rockenstein E, et al. Active immunization against α-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multisystem atrophy. Mol Neurodegen. 2015;10:721. doi:10.1186/s13024-015-0008-9
39. Schneeberger A, Tierney L, Mandler M. Active immunization therapies. Mov Disord. 2015;31(2):214-224. doi:10.1002/mds.26377
40. Zella SMA, Metzdorf J, Ciftci E, et al. Emerging immunotherapies for Parkinson disease. Neurol Ther. 2019;8(1):29-44. doi:10.1007/s40120-018-0122-z
41. AFFiRiS AG. AFFiRiS announces top line results of first-in-human clinical study using AFFITOPE PD03A, confirming immunogenicity and safety profile in Parkinson’s disease patients. https://affiris.com/wp-content/uploads/2018/10/praff011prefinal0607wo-embargo-1.pdf. Published June 7, 2017. Accessed July 29, 2020.
42. AFFiRiS AG. AFFiRiS announces results of a phase I clinical study using AFFITOPEs PD01A and PD03A, confirming safety and tolerability for both compounds as well as immunogenicity for PD01A in early MSA patients. http://sympath-project.eu/wp-content/uploads/PR_AFF009_V1.pdf Published March 1, 2018. Accessed July 29, 2020.
43. Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alphasynuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. doi:10.1371/journal.pone.0019338
44. Bae EJ, Lee HJ, Rockenstein E, et al. Antibody aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):1345-13469. doi:10.1523/JNEUROSCI.1292-12.2012
45. Schenk DB, Koller M, Ness DK, et al. First‐in‐human assessment of PRX002, an anti–α‐synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32(2):211-218. doi:10.1002/mds.26878.
46. Jankovic J, Goodman I, Safirstein B, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α -synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2018;75(10):1206-1214. doi:10.1001/jamaneurol.2018.1487
47. Jankovic J. Pathogenesis-targeted therapeutic strategies in Parkinson’s disease. Mov Disord. 2019;34(1):41-44. doi:10.1002/mds.27534
48. Shahaduzzaman M, Nash K, Hudson C, et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS One. 2015;10(2):E0116841. doi:10.1371/journal.pone.0116841
49. Brys M, Hung S, Fanning L, et al. Randomized, double-blind, placebo-controlled, single ascending dose study of anti-α-synuclein antibody BIIB054 in patients with Parkinson disease. Neurology. 2018;90(suppl 15):S26.001. doi:10.1002/mds.27738
50. Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target α-synuclein pathology. Exp Neurol. 2017;298(pt B):225-235. doi:10.1016/j.expneurol.2017.10.003
Parkinson disease (PD) is a progressive neurodegenerative disorder, characterized by diverse clinical symptoms. PD can present with rest tremor, bradykinesia, rigidity, falls, postural instability, and multiple nonmotor symptoms. Marras and colleagues estimated in a comprehensive meta-analysis that there were 680,000 individuals with PD in the US in 2010; this number is expected to double by 2030 based on the US Census Bureau population projections.1 An estimated 110,000 veterans may be affected by PD; hence, understanding of PD pathology, clinical progression, and effective treatment strategies is of paramount importance to the Veterans Health Administration (VHA).2
The exact pathogenesis underlying clinical features is still being studied. Pathologic diagnosis of PD relies on loss of dopamine neurons in the substantia nigra and accumulation of the abnormal protein, α-synuclein, in the form of Lewy bodies and Lewy neurites. Lewy bodies and neurites accumulate predominantly in the substantia nigra in addition to other brain stem nuclei and cerebral cortex. Lewy bodies are intraneuronal inclusions with a hyaline core and a pale peripheral halo. Central core stains positive for α-synuclein.3,4 Lewy neurites are widespread and are believed to play a larger role in the pathogenesis of PD compared with those of Lewy bodies.5
α-Synuclein
α-synuclein is a small 140 amino-acid protein with a N-terminal region that can interact with cell membranes and a highly acidic unstructured C-terminal region.6 α-synuclein is physiologically present in the presynaptic terminals of neurons and involved in synaptic plasticity and vesicle trafficking.7 There are different hypotheses about the native structure of α-synuclein. The first suggests that it exists in tetrameric form and may be broken down to monomer, which is the pathogenic form of α-synuclein. The second hypothesis suggests that it exists primarily in monomeric form, whereas other studies have shown that both forms exist and with pathologic changes, monomer accumulates in abundance and is neurotoxic.8-11 Work by Burré and colleagues shows that native α-synuclein exists in 2 forms: a soluble, cytosolic α-synuclein, which is monomeric, and a membrane-bound multimeric form.12,13
Alteration in aggregation properties of this protein is believed to play a central role in the pathogenesis of PD.14,15 Pathologic α-synuclein exists in insoluble forms that can aggregate into oligomers and fibrillar structures.16 Lysosomal dysfunction may promote accumulation of insoluble α-synuclein. Prior work has shown that several degradation pathways in lysosomes, including the ubiquitin-proteasome system and autophagy-lysosomal pathway, are down regulated, thus contributing to the accumulation of abnormal α-synuclein.17,18 Accumulation of pathologic α-synuclein leads to mitochondrial dysfunction in PD animal models, contributing further to neurotoxicity.19,20 Aggregates of phosphorylated α-synuclein have been demonstrated in dementia with Lewy body.21
In addition, α-synuclein aggregates may be released into extracellular spaces to be taken up by adjacent cells, where they can cause further misfolding and aggregation of protein.22 Previous work in animal models suggested a prion proteinlike spread of α-synuclein.23 This finding can have long-term therapeutic implications, as preventing extracellular release of abnormal form of α-synuclein will prevent the spread of pathologic protein. This can form the basis of neuroprotection in patients with PD.24
It has been proposed that α-synuclein accumulation and extracellular release initiates an immune response that leads to activation of microglia. This has been shown in PD animal models, overexpressing α-synuclein. In 2008 Park and colleagues demonstrated that microglial activation is enhanced by monomeric α-synuclein, not by the aggregated variant.25 Other studies have reported activated microglia around dopaminergic cells in substantia nigra.26 Sulzer and colleagues showed that peptides from α-synuclein can act as antigens and trigger an autoimmune reaction via T cells.27 PD may be associated with certain HLA-haplotypes.28 In other words, α-synuclein can induce neurodegeneration via different mechanisms, including alteration in synaptic vesicle transmission, mitochondrial dysfunction, neuroinflammation, and induction of humoral immunity.
Immunization
Due to these observations, there had been huge interest in developing antibody-based therapies for PD. A similar approach had been tested in Alzheimer disease (AD). Intracellular tangles of tau protein and extracellular aggregates of amyloid are the pathologic substrates in AD. Clinical trials utilizing antibodies targeting amyloid showed reduction in abnormal protein accumulation but no significant improvement in cognition.29 In addition, adverse events (AEs), such as vasogenic edema and intracerebral hemorrhage, were reported.30 Careful analysis of the data suggested that inadequate patient selection or targeting only amyloid, may have contributed to unfavorable results.31 Since then, more recent clinical trials have focused on careful patient selection, use of second generation anti-amyloid antibodies and immunotherapies targeting tau.32
Several studies have tested immunotherapies in PD animal models with the aim of targeting α-synuclein. Immunotherapies can be instituted in 2 ways: active immunization in which the immune system is stimulated to produce antibodies against α-synuclein or passive immunization in which antibodies against α-synuclein are administered directly. Once α-synuclein antibodies have crossed the blood-brain barrier, they are hypothesized to clear the existing α-synuclein. Animal studies have demonstrated the presence of these antibodies within the neurons. The mechanism of entry is unknown. Once inside the cells, the antibodies activate the lysosomal clearance, affecting intracellular accumulation of α-synuclein. Extracellularly, they can bind to receptors on scavenger cells, mainly microglia, activating them to facilitate uptake of extracellular α-synuclein. Binding of the antibodies to α-synuclein directly prevents the uptake of toxic protein by the cells, blocking the transfer and spread of PD pathology.33
Active Immunization
Active immunization against α-synuclein was demonstrated by Masliah and colleagues almost a decade ago. They administered recombinant human α-synuclein in transgenic mice expressing α-synuclein under the control of platelet-derived growth factor β. Reduction of accumulated α-synuclein in neurons with mild microglia activation was noted. It was proposed that the antibodies produced were able to bind to abnormal α-synuclein, were recognized by the lysosomal pathways, and degraded.34 Ghochikyan and colleagues developed vaccines by using α-synuclein-derived peptides. This induced formation of antibodies against α-synuclein in Lewy-bodies and neurites.35 Over time, other animal studies have been able to expand on these results.36
AFFiRiS, an Austrian biotechnology company, has developed 2 peptide vaccines PD01A and PD03A. Both peptides when administered to PD animal models caused antibody-based immune response against aggregated α-synuclein. Humoral autoimmune response was not observed in these studies; no neuroinflammation or neurotoxicity was noted. These peptides did not affect levels of physiologic α-synuclein, targeting only the aggregated form.37 These animal models showed improved motor and cognitive function. Similar results were noted in multiple system atrophy (MSA) animal models.38,39
The first human phase 1, randomized, parallel-group, single-center study recruited 32 subjects with early PD. Twelve subjects each were included in low- or high-dose treatment group, and 8 were included in the control group. Test subjects randomly received 4 vaccinations of low- or high-dose PD01A. Both doses were well tolerated, and no drug-related serious AEs were reported. The study confirmed the tolerability and safety of subcutaneous PD01A vaccine administration. These subjects were included in a 12-month, phase 1b follow-up extension study, AFF008E. In 2018, it was reported that administration of 6 doses of PD01A, 4 primary and 2 booster immunization, was safe. The vaccine showed a clear immune response against the peptide and cross-reactivity against α-synuclein targeted epitope. Booster doses stabilized the antibody titers. Significant increase in antibody titers against PD01A was seen over time, which was translated into a humoral immune response against α-synuclein. In addition, PD01A antibodies also were reported in cerebrospinal fluid.40
AFFiRiS presented results of a phase 1 randomized, placebo-controlled trial in 2017, confirming the safety of PD03A in patients with PD. The study showed a clear dose-dependent immune response against the peptide and cross-reactivity against α-synuclein targeted epitope.41 AFFiRiS recently presented results of another phase 1 clinical study assessing the safety and tolerability of vaccines PD01A and PD03A in patients with early MSA. Both vaccines were well tolerated, and PD01A induced an immune response against the peptide and α-synuclein epitope.42 These results have provided hope for further endeavors to develop active immunization strategies for PD.
Passive Immunization
Passive immunization against α-synuclein was first reported by Masliah and colleagues in 2011. A monoclonal antibody against the C-terminus of α-synuclein, 9E4, was injected into a transgenic mouse model of PD. There was reduction in α-synuclein aggregates in the brain along with improvement in motor and cognitive impairment.43 The C-terminus of α-synuclein plays a key role in the pathogenesis of PD. Changes in the C-terminus of α-synuclein induces formation of α-synuclein oligomers and subsequent neuronal spread. Antibody binds to the C-terminus and prevents structural changes that can lead to oligomerization of α-synuclein. Since the first study by Masliah, few other immunization studies utilized different antibodies against the C-terminus of α-synuclein. It was shown in a mouse model that binding of such antibodies promoted clearance of the α-synuclein by microglia.44
Based on these animal studies, Prothena Biosciences (South San Francisco, CA) designed a phase 1, double-blind, randomized, placebo-controlled clinical trial of prasinezumab (investigational monoclonal antibody against C-terminus of α-synuclein), in subjects without PD. The results showed that it was well tolerated, and there was dose-dependent reduction in the levels of free
BIIB054 is another monoclonal antibody that targets the N-terminal of α-synuclein. In animal models, antibodies targeting the N-terminus reduced α-synuclein triggered cell death and reduced the number of activated microglia.48 BIIB054, from Biogen (Cambridge, MA), was studied in 40 healthy subjects and was well tolerated with a favorable safety profile and could cross the blood-brain barrier. Like the prasinezumab study, this also was an ascending-dose study to assess safety and tolerability. In 2018, a randomized, double-blind, placebo-controlled, single-ascending dose study in patients with PD reported that BIIB054 was well tolerated, and the presence of BIIB054-synuclein complexes in the plasma were confirmed.49 A phase 2, multicenter, randomized, double-blind, placebo-controlled study (SPARK) with an active-treatment dose-blinded period, designed to evaluate the safety, pharmacokinetics, and the pharmacodynamics of BIIB054 is currently recruiting patients with PD.
Finally, BioArctic (Stockholm, Sweden) developed antibodies that are selective for oligomeric forms of α-synuclein, which it licensed to AbbVie (North Chicago, Il).50 These antibodies do not target the N- or C-terminus of α-synuclein. Since α-synuclein oligomers play an important role in the pathogenesis of PD, targeting them with antibodies at an early stage may prove to be an effective strategy for removal of pathogenic α-synuclein. Clinical trials are forthcoming.
Conclusions
Immunotherapy against α-synuclein has provided a new therapeutic avenue in the field of neuroprotection. Results from the first human clinical trial are promising, but despite these results, more work is needed to clarify the role of α-synuclein in the pathogenesis of PD in humans. Most of the work concerning α-synuclein aggregation and propagation has been reported in animal models. Whether similar process exists in humans is a debatable question. Similarly, more knowledge is needed about how and where in the human brain antibodies act to give neuroprotective effects. Timing of administration of immunotherapies in real time will be a crucial question.
PD is clinically evident once 80% of dopaminergic neurons in substantia nigra are lost due to neurodegeneration. Should immunotherapy be administered to symptomatic patients with PD, or if it will be beneficial only for presymptomatic, high-risk patients needs to be determined. Like AD trials, not only careful selection of patients, but determination of optimal timing for treatment will be essential. As the understanding of PD pathogenesis and therapeutics evolves, it will become clear whether immunization targeting α-synuclein will modify disease progression.
Parkinson disease (PD) is a progressive neurodegenerative disorder, characterized by diverse clinical symptoms. PD can present with rest tremor, bradykinesia, rigidity, falls, postural instability, and multiple nonmotor symptoms. Marras and colleagues estimated in a comprehensive meta-analysis that there were 680,000 individuals with PD in the US in 2010; this number is expected to double by 2030 based on the US Census Bureau population projections.1 An estimated 110,000 veterans may be affected by PD; hence, understanding of PD pathology, clinical progression, and effective treatment strategies is of paramount importance to the Veterans Health Administration (VHA).2
The exact pathogenesis underlying clinical features is still being studied. Pathologic diagnosis of PD relies on loss of dopamine neurons in the substantia nigra and accumulation of the abnormal protein, α-synuclein, in the form of Lewy bodies and Lewy neurites. Lewy bodies and neurites accumulate predominantly in the substantia nigra in addition to other brain stem nuclei and cerebral cortex. Lewy bodies are intraneuronal inclusions with a hyaline core and a pale peripheral halo. Central core stains positive for α-synuclein.3,4 Lewy neurites are widespread and are believed to play a larger role in the pathogenesis of PD compared with those of Lewy bodies.5
α-Synuclein
α-synuclein is a small 140 amino-acid protein with a N-terminal region that can interact with cell membranes and a highly acidic unstructured C-terminal region.6 α-synuclein is physiologically present in the presynaptic terminals of neurons and involved in synaptic plasticity and vesicle trafficking.7 There are different hypotheses about the native structure of α-synuclein. The first suggests that it exists in tetrameric form and may be broken down to monomer, which is the pathogenic form of α-synuclein. The second hypothesis suggests that it exists primarily in monomeric form, whereas other studies have shown that both forms exist and with pathologic changes, monomer accumulates in abundance and is neurotoxic.8-11 Work by Burré and colleagues shows that native α-synuclein exists in 2 forms: a soluble, cytosolic α-synuclein, which is monomeric, and a membrane-bound multimeric form.12,13
Alteration in aggregation properties of this protein is believed to play a central role in the pathogenesis of PD.14,15 Pathologic α-synuclein exists in insoluble forms that can aggregate into oligomers and fibrillar structures.16 Lysosomal dysfunction may promote accumulation of insoluble α-synuclein. Prior work has shown that several degradation pathways in lysosomes, including the ubiquitin-proteasome system and autophagy-lysosomal pathway, are down regulated, thus contributing to the accumulation of abnormal α-synuclein.17,18 Accumulation of pathologic α-synuclein leads to mitochondrial dysfunction in PD animal models, contributing further to neurotoxicity.19,20 Aggregates of phosphorylated α-synuclein have been demonstrated in dementia with Lewy body.21
In addition, α-synuclein aggregates may be released into extracellular spaces to be taken up by adjacent cells, where they can cause further misfolding and aggregation of protein.22 Previous work in animal models suggested a prion proteinlike spread of α-synuclein.23 This finding can have long-term therapeutic implications, as preventing extracellular release of abnormal form of α-synuclein will prevent the spread of pathologic protein. This can form the basis of neuroprotection in patients with PD.24
It has been proposed that α-synuclein accumulation and extracellular release initiates an immune response that leads to activation of microglia. This has been shown in PD animal models, overexpressing α-synuclein. In 2008 Park and colleagues demonstrated that microglial activation is enhanced by monomeric α-synuclein, not by the aggregated variant.25 Other studies have reported activated microglia around dopaminergic cells in substantia nigra.26 Sulzer and colleagues showed that peptides from α-synuclein can act as antigens and trigger an autoimmune reaction via T cells.27 PD may be associated with certain HLA-haplotypes.28 In other words, α-synuclein can induce neurodegeneration via different mechanisms, including alteration in synaptic vesicle transmission, mitochondrial dysfunction, neuroinflammation, and induction of humoral immunity.
Immunization
Due to these observations, there had been huge interest in developing antibody-based therapies for PD. A similar approach had been tested in Alzheimer disease (AD). Intracellular tangles of tau protein and extracellular aggregates of amyloid are the pathologic substrates in AD. Clinical trials utilizing antibodies targeting amyloid showed reduction in abnormal protein accumulation but no significant improvement in cognition.29 In addition, adverse events (AEs), such as vasogenic edema and intracerebral hemorrhage, were reported.30 Careful analysis of the data suggested that inadequate patient selection or targeting only amyloid, may have contributed to unfavorable results.31 Since then, more recent clinical trials have focused on careful patient selection, use of second generation anti-amyloid antibodies and immunotherapies targeting tau.32
Several studies have tested immunotherapies in PD animal models with the aim of targeting α-synuclein. Immunotherapies can be instituted in 2 ways: active immunization in which the immune system is stimulated to produce antibodies against α-synuclein or passive immunization in which antibodies against α-synuclein are administered directly. Once α-synuclein antibodies have crossed the blood-brain barrier, they are hypothesized to clear the existing α-synuclein. Animal studies have demonstrated the presence of these antibodies within the neurons. The mechanism of entry is unknown. Once inside the cells, the antibodies activate the lysosomal clearance, affecting intracellular accumulation of α-synuclein. Extracellularly, they can bind to receptors on scavenger cells, mainly microglia, activating them to facilitate uptake of extracellular α-synuclein. Binding of the antibodies to α-synuclein directly prevents the uptake of toxic protein by the cells, blocking the transfer and spread of PD pathology.33
Active Immunization
Active immunization against α-synuclein was demonstrated by Masliah and colleagues almost a decade ago. They administered recombinant human α-synuclein in transgenic mice expressing α-synuclein under the control of platelet-derived growth factor β. Reduction of accumulated α-synuclein in neurons with mild microglia activation was noted. It was proposed that the antibodies produced were able to bind to abnormal α-synuclein, were recognized by the lysosomal pathways, and degraded.34 Ghochikyan and colleagues developed vaccines by using α-synuclein-derived peptides. This induced formation of antibodies against α-synuclein in Lewy-bodies and neurites.35 Over time, other animal studies have been able to expand on these results.36
AFFiRiS, an Austrian biotechnology company, has developed 2 peptide vaccines PD01A and PD03A. Both peptides when administered to PD animal models caused antibody-based immune response against aggregated α-synuclein. Humoral autoimmune response was not observed in these studies; no neuroinflammation or neurotoxicity was noted. These peptides did not affect levels of physiologic α-synuclein, targeting only the aggregated form.37 These animal models showed improved motor and cognitive function. Similar results were noted in multiple system atrophy (MSA) animal models.38,39
The first human phase 1, randomized, parallel-group, single-center study recruited 32 subjects with early PD. Twelve subjects each were included in low- or high-dose treatment group, and 8 were included in the control group. Test subjects randomly received 4 vaccinations of low- or high-dose PD01A. Both doses were well tolerated, and no drug-related serious AEs were reported. The study confirmed the tolerability and safety of subcutaneous PD01A vaccine administration. These subjects were included in a 12-month, phase 1b follow-up extension study, AFF008E. In 2018, it was reported that administration of 6 doses of PD01A, 4 primary and 2 booster immunization, was safe. The vaccine showed a clear immune response against the peptide and cross-reactivity against α-synuclein targeted epitope. Booster doses stabilized the antibody titers. Significant increase in antibody titers against PD01A was seen over time, which was translated into a humoral immune response against α-synuclein. In addition, PD01A antibodies also were reported in cerebrospinal fluid.40
AFFiRiS presented results of a phase 1 randomized, placebo-controlled trial in 2017, confirming the safety of PD03A in patients with PD. The study showed a clear dose-dependent immune response against the peptide and cross-reactivity against α-synuclein targeted epitope.41 AFFiRiS recently presented results of another phase 1 clinical study assessing the safety and tolerability of vaccines PD01A and PD03A in patients with early MSA. Both vaccines were well tolerated, and PD01A induced an immune response against the peptide and α-synuclein epitope.42 These results have provided hope for further endeavors to develop active immunization strategies for PD.
Passive Immunization
Passive immunization against α-synuclein was first reported by Masliah and colleagues in 2011. A monoclonal antibody against the C-terminus of α-synuclein, 9E4, was injected into a transgenic mouse model of PD. There was reduction in α-synuclein aggregates in the brain along with improvement in motor and cognitive impairment.43 The C-terminus of α-synuclein plays a key role in the pathogenesis of PD. Changes in the C-terminus of α-synuclein induces formation of α-synuclein oligomers and subsequent neuronal spread. Antibody binds to the C-terminus and prevents structural changes that can lead to oligomerization of α-synuclein. Since the first study by Masliah, few other immunization studies utilized different antibodies against the C-terminus of α-synuclein. It was shown in a mouse model that binding of such antibodies promoted clearance of the α-synuclein by microglia.44
Based on these animal studies, Prothena Biosciences (South San Francisco, CA) designed a phase 1, double-blind, randomized, placebo-controlled clinical trial of prasinezumab (investigational monoclonal antibody against C-terminus of α-synuclein), in subjects without PD. The results showed that it was well tolerated, and there was dose-dependent reduction in the levels of free
BIIB054 is another monoclonal antibody that targets the N-terminal of α-synuclein. In animal models, antibodies targeting the N-terminus reduced α-synuclein triggered cell death and reduced the number of activated microglia.48 BIIB054, from Biogen (Cambridge, MA), was studied in 40 healthy subjects and was well tolerated with a favorable safety profile and could cross the blood-brain barrier. Like the prasinezumab study, this also was an ascending-dose study to assess safety and tolerability. In 2018, a randomized, double-blind, placebo-controlled, single-ascending dose study in patients with PD reported that BIIB054 was well tolerated, and the presence of BIIB054-synuclein complexes in the plasma were confirmed.49 A phase 2, multicenter, randomized, double-blind, placebo-controlled study (SPARK) with an active-treatment dose-blinded period, designed to evaluate the safety, pharmacokinetics, and the pharmacodynamics of BIIB054 is currently recruiting patients with PD.
Finally, BioArctic (Stockholm, Sweden) developed antibodies that are selective for oligomeric forms of α-synuclein, which it licensed to AbbVie (North Chicago, Il).50 These antibodies do not target the N- or C-terminus of α-synuclein. Since α-synuclein oligomers play an important role in the pathogenesis of PD, targeting them with antibodies at an early stage may prove to be an effective strategy for removal of pathogenic α-synuclein. Clinical trials are forthcoming.
Conclusions
Immunotherapy against α-synuclein has provided a new therapeutic avenue in the field of neuroprotection. Results from the first human clinical trial are promising, but despite these results, more work is needed to clarify the role of α-synuclein in the pathogenesis of PD in humans. Most of the work concerning α-synuclein aggregation and propagation has been reported in animal models. Whether similar process exists in humans is a debatable question. Similarly, more knowledge is needed about how and where in the human brain antibodies act to give neuroprotective effects. Timing of administration of immunotherapies in real time will be a crucial question.
PD is clinically evident once 80% of dopaminergic neurons in substantia nigra are lost due to neurodegeneration. Should immunotherapy be administered to symptomatic patients with PD, or if it will be beneficial only for presymptomatic, high-risk patients needs to be determined. Like AD trials, not only careful selection of patients, but determination of optimal timing for treatment will be essential. As the understanding of PD pathogenesis and therapeutics evolves, it will become clear whether immunization targeting α-synuclein will modify disease progression.
1. Marras C, Beck JC, Bower JH, et al; Parkinson’s Foundation P4 Group. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018;4(1):21. doi:10.1038/s41531-018-0058-0
2. Mantri S, Duda JE, Morley JF. Early and accurate identification of Parkinson disease among US veterans. Fed Pract. 2019;36(suppl 4):S18-S23. doi:10.12788/fp.37-0034
3. Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7(suppl 1):S71-S85. doi:10.3233/JPD-179001
4. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839-840. doi:10.1038/42166
5. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197-211. doi:10.1016/s0197-4580(02)00065-9
6. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79(6):1044-1066. doi:10.1016/j.neuron.2013.09.004
7. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663-1667. doi:10.1126/science.1195227
8. Binolfi A, Fernández CO, Sica MP, Delfino JM, Santos J. Recognition between a short unstructured peptide and a partially folded fragment leads to the thioredoxin fold sharing native-like dynamics. Proteins. 2012;80(5):1448-1464. doi:10.1002/prot.24043
9. Fauvet B, Mbefo MK, Fares MB, et al. α-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345-15364. doi:10.1074/jbc.M111.318949.
10. Wang W, Perovic I, Chittuluru J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108(43):17797-17802. doi:10.1073/pnas.1113260108
11. Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P. From α-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012;1476:183-202. doi:10.1016/j.brainres.2012.04.014
12. Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native α-synuclein. Nature. 2013;498(7453):E4-E7.
13. Burré J, Sharma M, Südhof TC. α-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA. 2014;111(40):E4274-E4283. doi:10.1073/pnas.1416598111
14. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1-13. doi:10.1038/nm.4269
15. Burré J, Sharma M, Südhof TC. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci. 2015;35(13):5221-5232. doi:10.1523/JNEUROSCI.4650-14.2015
16. Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24(8):1888-1896. doi:10.1523/JNEUROSCI.3809-03.2004
17. Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L . α-synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem. 2004;279(45):46915-46920. doi:10.1074/jbc.M405146200
18. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200-210. doi:10.1016/j.tibs.2015.02.003
19. Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochem Biophys Acta. 2010;1802(1):29-44. doi:10.1016/j.bbadis.2009.08.013
20. Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein: a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92-98. doi:10.1038/nrneurol.2013.275
21. Colom-Cadena M, Pegueroles J, Herrmann AG, et al. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain. 2017;140(12):3204-3214. doi:10.1093/brain/awx275
22. Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57-71. doi:10.1016/j.neuron.2011.08.033
23. Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(pt 4):1128-1138. doi:10.1093/brain/awt037
24. Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22-33. doi:10.1016/j.pharmthera.2016.11.010
25. Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56(11):1215-1223. doi:10.1002/glia.20691
26. Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimmune Pharmacol. 2013;8(1):189-201. doi:10.1007/s11481-013-9435-y
27. Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546(7660):656-661. doi:10.1038/nature22815
28. Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genetics. 2010;42(9):781-785. doi:10.1038/ng.642
29. Holmes C, Boche D, Wilkinson D, et al. Long term effects of Aβ42 immunisation in Alzheimer’s disease: follow up of a randomized, placebo-controlled phase I trial. Lancet. 2008;372(9634):216-223. doi:10.1016/S0140-6736(08)61075-2
30. Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241-249. doi:10.1016/S1474-4422(12)70015-7
31. Wisniewski T, Goñi F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499-507. doi:10.1016/j.bcp.2013.12.020
32. Herline K, Drummond E, Wisniewski T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines. 2018;17(8):707-721. doi:10.1080/14760584.2018.1500905
33. Bergstrom AL, Kallunki P, Fog K. Development of passive immunotherapies for synucleopathies. Mov Disord. 2015;31(2):203-213. doi:10.1002/mds.26481
34. Masliah E, Rockenstein E, Adame A, et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857-868. doi:10.1016/j.neuron.2005.05.010
35. Ghochikyan A, Petrushina I, Davtyan H, et al. Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: feasibility study. Neurosci Lett. 2014;560:86-91. doi:10.1016/j.neulet.2013.12.028
36. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. α-synuclein vaccination prevents the accumulation of Parkinson’s disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72(7):624-645. doi:10.1097/NEN.0b013e31829768d2
37. Mandler M, Valera E, Rockenstein E, et al. Next generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127(6):861-879. doi:10.1007/s00401-014-1256-4
38. Mandler M, Valera E, Rockenstein E, et al. Active immunization against α-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multisystem atrophy. Mol Neurodegen. 2015;10:721. doi:10.1186/s13024-015-0008-9
39. Schneeberger A, Tierney L, Mandler M. Active immunization therapies. Mov Disord. 2015;31(2):214-224. doi:10.1002/mds.26377
40. Zella SMA, Metzdorf J, Ciftci E, et al. Emerging immunotherapies for Parkinson disease. Neurol Ther. 2019;8(1):29-44. doi:10.1007/s40120-018-0122-z
41. AFFiRiS AG. AFFiRiS announces top line results of first-in-human clinical study using AFFITOPE PD03A, confirming immunogenicity and safety profile in Parkinson’s disease patients. https://affiris.com/wp-content/uploads/2018/10/praff011prefinal0607wo-embargo-1.pdf. Published June 7, 2017. Accessed July 29, 2020.
42. AFFiRiS AG. AFFiRiS announces results of a phase I clinical study using AFFITOPEs PD01A and PD03A, confirming safety and tolerability for both compounds as well as immunogenicity for PD01A in early MSA patients. http://sympath-project.eu/wp-content/uploads/PR_AFF009_V1.pdf Published March 1, 2018. Accessed July 29, 2020.
43. Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alphasynuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. doi:10.1371/journal.pone.0019338
44. Bae EJ, Lee HJ, Rockenstein E, et al. Antibody aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):1345-13469. doi:10.1523/JNEUROSCI.1292-12.2012
45. Schenk DB, Koller M, Ness DK, et al. First‐in‐human assessment of PRX002, an anti–α‐synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32(2):211-218. doi:10.1002/mds.26878.
46. Jankovic J, Goodman I, Safirstein B, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α -synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2018;75(10):1206-1214. doi:10.1001/jamaneurol.2018.1487
47. Jankovic J. Pathogenesis-targeted therapeutic strategies in Parkinson’s disease. Mov Disord. 2019;34(1):41-44. doi:10.1002/mds.27534
48. Shahaduzzaman M, Nash K, Hudson C, et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS One. 2015;10(2):E0116841. doi:10.1371/journal.pone.0116841
49. Brys M, Hung S, Fanning L, et al. Randomized, double-blind, placebo-controlled, single ascending dose study of anti-α-synuclein antibody BIIB054 in patients with Parkinson disease. Neurology. 2018;90(suppl 15):S26.001. doi:10.1002/mds.27738
50. Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target α-synuclein pathology. Exp Neurol. 2017;298(pt B):225-235. doi:10.1016/j.expneurol.2017.10.003
1. Marras C, Beck JC, Bower JH, et al; Parkinson’s Foundation P4 Group. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018;4(1):21. doi:10.1038/s41531-018-0058-0
2. Mantri S, Duda JE, Morley JF. Early and accurate identification of Parkinson disease among US veterans. Fed Pract. 2019;36(suppl 4):S18-S23. doi:10.12788/fp.37-0034
3. Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7(suppl 1):S71-S85. doi:10.3233/JPD-179001
4. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839-840. doi:10.1038/42166
5. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197-211. doi:10.1016/s0197-4580(02)00065-9
6. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79(6):1044-1066. doi:10.1016/j.neuron.2013.09.004
7. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663-1667. doi:10.1126/science.1195227
8. Binolfi A, Fernández CO, Sica MP, Delfino JM, Santos J. Recognition between a short unstructured peptide and a partially folded fragment leads to the thioredoxin fold sharing native-like dynamics. Proteins. 2012;80(5):1448-1464. doi:10.1002/prot.24043
9. Fauvet B, Mbefo MK, Fares MB, et al. α-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345-15364. doi:10.1074/jbc.M111.318949.
10. Wang W, Perovic I, Chittuluru J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108(43):17797-17802. doi:10.1073/pnas.1113260108
11. Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P. From α-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012;1476:183-202. doi:10.1016/j.brainres.2012.04.014
12. Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native α-synuclein. Nature. 2013;498(7453):E4-E7.
13. Burré J, Sharma M, Südhof TC. α-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA. 2014;111(40):E4274-E4283. doi:10.1073/pnas.1416598111
14. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1-13. doi:10.1038/nm.4269
15. Burré J, Sharma M, Südhof TC. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci. 2015;35(13):5221-5232. doi:10.1523/JNEUROSCI.4650-14.2015
16. Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24(8):1888-1896. doi:10.1523/JNEUROSCI.3809-03.2004
17. Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L . α-synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem. 2004;279(45):46915-46920. doi:10.1074/jbc.M405146200
18. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200-210. doi:10.1016/j.tibs.2015.02.003
19. Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochem Biophys Acta. 2010;1802(1):29-44. doi:10.1016/j.bbadis.2009.08.013
20. Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein: a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92-98. doi:10.1038/nrneurol.2013.275
21. Colom-Cadena M, Pegueroles J, Herrmann AG, et al. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain. 2017;140(12):3204-3214. doi:10.1093/brain/awx275
22. Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57-71. doi:10.1016/j.neuron.2011.08.033
23. Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(pt 4):1128-1138. doi:10.1093/brain/awt037
24. Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22-33. doi:10.1016/j.pharmthera.2016.11.010
25. Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56(11):1215-1223. doi:10.1002/glia.20691
26. Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimmune Pharmacol. 2013;8(1):189-201. doi:10.1007/s11481-013-9435-y
27. Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546(7660):656-661. doi:10.1038/nature22815
28. Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genetics. 2010;42(9):781-785. doi:10.1038/ng.642
29. Holmes C, Boche D, Wilkinson D, et al. Long term effects of Aβ42 immunisation in Alzheimer’s disease: follow up of a randomized, placebo-controlled phase I trial. Lancet. 2008;372(9634):216-223. doi:10.1016/S0140-6736(08)61075-2
30. Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241-249. doi:10.1016/S1474-4422(12)70015-7
31. Wisniewski T, Goñi F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499-507. doi:10.1016/j.bcp.2013.12.020
32. Herline K, Drummond E, Wisniewski T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines. 2018;17(8):707-721. doi:10.1080/14760584.2018.1500905
33. Bergstrom AL, Kallunki P, Fog K. Development of passive immunotherapies for synucleopathies. Mov Disord. 2015;31(2):203-213. doi:10.1002/mds.26481
34. Masliah E, Rockenstein E, Adame A, et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857-868. doi:10.1016/j.neuron.2005.05.010
35. Ghochikyan A, Petrushina I, Davtyan H, et al. Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: feasibility study. Neurosci Lett. 2014;560:86-91. doi:10.1016/j.neulet.2013.12.028
36. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. α-synuclein vaccination prevents the accumulation of Parkinson’s disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72(7):624-645. doi:10.1097/NEN.0b013e31829768d2
37. Mandler M, Valera E, Rockenstein E, et al. Next generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127(6):861-879. doi:10.1007/s00401-014-1256-4
38. Mandler M, Valera E, Rockenstein E, et al. Active immunization against α-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multisystem atrophy. Mol Neurodegen. 2015;10:721. doi:10.1186/s13024-015-0008-9
39. Schneeberger A, Tierney L, Mandler M. Active immunization therapies. Mov Disord. 2015;31(2):214-224. doi:10.1002/mds.26377
40. Zella SMA, Metzdorf J, Ciftci E, et al. Emerging immunotherapies for Parkinson disease. Neurol Ther. 2019;8(1):29-44. doi:10.1007/s40120-018-0122-z
41. AFFiRiS AG. AFFiRiS announces top line results of first-in-human clinical study using AFFITOPE PD03A, confirming immunogenicity and safety profile in Parkinson’s disease patients. https://affiris.com/wp-content/uploads/2018/10/praff011prefinal0607wo-embargo-1.pdf. Published June 7, 2017. Accessed July 29, 2020.
42. AFFiRiS AG. AFFiRiS announces results of a phase I clinical study using AFFITOPEs PD01A and PD03A, confirming safety and tolerability for both compounds as well as immunogenicity for PD01A in early MSA patients. http://sympath-project.eu/wp-content/uploads/PR_AFF009_V1.pdf Published March 1, 2018. Accessed July 29, 2020.
43. Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alphasynuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. doi:10.1371/journal.pone.0019338
44. Bae EJ, Lee HJ, Rockenstein E, et al. Antibody aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):1345-13469. doi:10.1523/JNEUROSCI.1292-12.2012
45. Schenk DB, Koller M, Ness DK, et al. First‐in‐human assessment of PRX002, an anti–α‐synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32(2):211-218. doi:10.1002/mds.26878.
46. Jankovic J, Goodman I, Safirstein B, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α -synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2018;75(10):1206-1214. doi:10.1001/jamaneurol.2018.1487
47. Jankovic J. Pathogenesis-targeted therapeutic strategies in Parkinson’s disease. Mov Disord. 2019;34(1):41-44. doi:10.1002/mds.27534
48. Shahaduzzaman M, Nash K, Hudson C, et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS One. 2015;10(2):E0116841. doi:10.1371/journal.pone.0116841
49. Brys M, Hung S, Fanning L, et al. Randomized, double-blind, placebo-controlled, single ascending dose study of anti-α-synuclein antibody BIIB054 in patients with Parkinson disease. Neurology. 2018;90(suppl 15):S26.001. doi:10.1002/mds.27738
50. Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target α-synuclein pathology. Exp Neurol. 2017;298(pt B):225-235. doi:10.1016/j.expneurol.2017.10.003
The Use of Immuno-Oncology Treatments in the VA (FULL)
The following is a lightly edited transcript of a teleconference discussion recorded in April 2018.
Suman Kambhampati, MD. Immuno-oncology is a paradigm-shifting treatment approach. It is an easy-to-understand term for both providers and for patients. The underlying principle is that the body’s own immune system is used or stimulated to fight cancer, and there are drugs that clearly have shown huge promise for this, not only in oncology, but also for other diseases. Time will tell whether that really pans out or not, but to begin with, the emphasis has been inoncology, and therefore, the term immunooncology is fitting.
Dr. Kaster. It was encouraging at first, especially when ipilimumab came out, to see the effects on patients with melanoma. Then the KEYNOTE-024 trial came out, and we were able to jump in anduse monoclonal antibodies directed against programmed death 1 (PD-1) in the first line, which is when things got exciting.1 We have a smaller populationin Boise, so PD-1s in lung cancer have had the biggest impact on our patients so far.
Ellen Nason, RN, MSN. Patients are open to immunotherapies.They’re excited about it. And as the other panelists have said, you can start broadly, as the body fights the cancer on its own, to providing more specific details as a patient wants more information. Immuno-oncology is definitely accepted by patients, and they’re very excited about it, especially with all the news about new therapies.
Dr. Kambhampati. For the Department of Veteran Affairs (VA) population, lung cancer has seen significant impact, and now it’s translating into other diseases through more research, trials, and better understanding about how these drugs are used and work. 
The paradigm is shifting toward offering these drugs not only in metastatic cancers, but also in the surgically resectable tumors. The 2018 American Association for Cancer Research (AACR) meeting, just concluded. At the meeting several abstracts reported instances where immunooncology drugs are being introduced in the early phases of lung cancer and showing outstanding results. It’s very much possible that we’re going to see less use of traditional chemotherapy in the near future.
Ms. Nason. I primarily work with solid tumors,and the majority of the population I work with have lung cancer. So we’re excited about some of the results that we’ve seen and the lower toxicity involved. Recently, we’ve begun using durvalumab with patients with stage III disease. We have about 5 people now that are using it as a maintenance or consolidative treatment vs just using it for patients with stage IV disease. Hopefully, we’ll see some of the same results describedin the paper published on it.2
Dr. Kaster. Yes, we are incorporating these new changes into care as they're coming out. As Ms. Nason mentioned, we're already using immunotherapies in earlier settings, and we are seeing as much research that could be translated into care soon, like combining immunotherapies
in first-line settings, as we see in the Checkmate-227 study with nivolumab and ipilimumab.3,4 The landscape is going to change dramatically in the next couple of years.
Accessing Testing For First-Line Treatments
Dr. Lynch. There has been an ongoing discussionin the literature on accessing appropriate testing—delays in testing can result in patients who are not able to access the best targeted drugs on a first-line basis. The drug companiesand the VA have become highly sensitized to ensuring that veterans are accessing the appropriate testing. We are expanding the capability of VA labs to do that testing.
Ms. Nason. I want to put in a plug for the VA Precision Oncology Program (POP). It’s about 2 years into its existence, and Neil Spector, MD, is the director. The POP pays for sequencing the tumor samples.
A new sequencing contract will go into effect October 2018 and will include sequencing for hematologic malignancies in addition to the current testing of solid tumors. Patients from New York who have been unable to receive testing through the current vendors used by POP, will be included in the new contract. It is important to note that POP is working closely with the National Pharmacy Benefit Management Service (PBM) to develop a policy for approving off-label use of US Food and Drug Administration-approved targeted therapies based on sequenced data collected on patients tested through POP.
In addition, the leadership of POP is working to leverage the molecular testing results conducted through POP to improve veterans' access to clinical trials, both inside and outside the VA. Within the VA people can access information at tinyurl.com/precisiononcology. There is no reason why any eligible patient with cancer in the VA health care system should not have their tumor tissue sequenced through POP, particularly once the new contract goes into effect.
Dr. Lynch. Fortunately, the cost of next-generation sequencing has come down so much that most VA contracted reference laboratories offer next-generation sequencing, including LabCorp (Burlington,NC), Quest Diagnostics (Secaucus, NJ), Fulgent (Temple City, CA), and academic partners such as Oregon Health Sciences University and University of Washington.
Ms. Nason. At the Durham VAMC, sometimes a lack of tissue has been a barrier, but we now have the ability to send blood (liquid biopsy) for next-generation sequencing. Hopefully that will open up options for veterans with inadequate tissue. Importantly, all VA facilities can request liquid biopsiesthrough POP.
Dr. Lynch. That’s an important point. There have been huge advances in liquid biopsy testing.The VA Salt Lake City Health Care System (VASLCHCS) was in talks with Genomic Health (Redwood City, CA) to do a study as part of clinical operations to look at the concordance between the liquid biopsy testing and the precision oncology data. But Genomic Health eventually abandoned its liquid biopsy testing. Currently, the VA is only reimbursing or encouraging liquid biopsy if the tissue is not available or if the veteran has too high a level of comorbidities to undergo tissue biopsy. The main point for the discussion today is that access to testing is a key component of access to all of these advanced drugs.
Dr. Kambhampati. The precision medicine piece will be a game changer—no question about that. Liquid biopsy is very timely. Many patients have difficulty getting rebiopsied, so liquid biopsy is definitely a big, big step forward.
Still, there has not been consistency across the VA as there should be. Perhaps there are a few select centers, including our site in Kansas City, where access to precision medicine is readily available and liquid biopsies are available. We use the PlasmaSELECT test from Personal Genome Diagnostics (Baltimore, MD). We have just added Foundation Medicine (Cambridge, MA) also in hematology. Access to mutational profilingis absolutely a must for precision medicine.
All that being said, the unique issue with immuno-oncology is that it pretty much transcends the mutational profile and perhaps has leveled the playing field, irrespective of the tumor mutation profile or burden. In some solid tumors these immuno-oncology drugs have been shown to work across tumor types and across different mutation types. And there is a hint now in the recent data presented at AACR and in the New England Journalof Medicine showing that the tumor mutational burden is a predictor of pathologic response to at least PD-1 blockade in the resectable stages of lung cancer.1,3 To me, that’s a very important piece of data because that’s something that can be tested and can have a prognostic impact in immuno-oncology, particularly in the early stages of lung cancer and is further proof of the broad value of immunotherapics in targeting tumors irrespective of the precise tumor targets.
Dr. Kaster. Yes, it’s nice to see other options like tumor mutational burden and Lung Immune Prognostic Index being studied.5 It would be nice if we could rely a little more on these, and not PD-L1, which as we all know is a variable and an unreliable target.
Dr. Kambhampati. I agree.
Rural Challenges In A Veterans Population
Dr. Lynch. Providing high-quality cancer care to rural veterans care can be a challenge but it is a VA priority. The VA National Genomic Medicine Services offers better access for rural veterans to germline genetic testing than any other healthcare system in the country. In terms of access to somatic testing and next-generation sequencing, we are working toward providing the same level of cancer care as patients would receive at National Cancer Institute (NCI) cancer centers. The VA oncology leadership has done teleconsults and virtual tumor boards, but for some rural VAMCs, fellowsare leading the clinical care. As we expand use of oral agents for oncology treatment, it will be easier to ensure that rural veterans receive the same standard of care for POP that veterans being cared for at VASLCHCS, Kansas City VAMC, or Durham VAMC get.
Dr. Kambhampati. The Kansas City VAMC in its catchment area includes underserved areas, such as Topeka and Leavenworth, Kansas. What we’ve been able to do here is something that’s unique—Kansas City VAMC is the only standalone VA in the country to be recognized as a primary SWOG (Southwestern Oncology Group) institution, which provides access to many trials, such as the Lung-MAP trial and others. And that has allowed us to use the full expanse of precision medicine without financial barriers. The research has helped us improve the standard of
care for patients across VISN 15.
Dr. Lynch. In precision oncology, the chief of pathology is an important figure in access to advanced care. I’ve worked with Sharad Mathur,MD, of the Kansas City VAMC on many clinical trials. He’s on the Kansas City VAMC Institutional Review Board and the cancer committee and is tuned in to veterans’ access to precision oncology. Kansas City was ordering Foundation One for select patients that met the criteria probably sooner than any other VA and participated in NCI Cooperative Group clinical trials. It is a great example of how veterans are getting access to
the same level of care as are patients who gettreated at NCI partners.
Comorbidities
Dr. Kambhampati. I don’t treat a lot of patients with lung cancer, but I find it easier to use these immuno-oncology drugs than platinums and etoposide. I consider them absolutely nasty chemotherapy drugs now in this era of immuno-oncology and targeted therapy.
Dr. Lynch. The VA is very important in translational lung cancer research and clinical care. It used to be thought that African American patients don’t get epidermal growth factor receptor mutations. And that’s because not enough African American patients with lung cancer were included in the NCI-based clinical trial.There are7,000 veterans who get lung cancer each year, and 20% to 25% of those are African Americans. Prevalence of various mutations and the pharmacogenetics of some of these drugs differ by patient ancestry. Including veterans with lung
cancer in precision oncology clinical trials and clinical care is not just a priority for the VA but a priority for NCI and internationally. I can’t emphasize this enough—veterans with lung cancer should be included in these studies and should be getting the same level of care that our partners are getting at NCI cancer centers. In the VA we’re positioned to do this because of our nationalelectronic health record (EHR) and becauseof our ability to identify patients with specific variants and enroll them in clinical trials.
Ms. Nason. One of the barriers that I find withsome of the patients that I have treated is getting them to a trial. If the trial isn’t available locally, specifically there are socioeconomic and distance issues that are hard to overcome.
Dr. Kaster. For smaller medical centers, getting patients to clinical trials can be difficult. The Boise VAMC is putting together a proposal now to justify hiring a research pharmacist in order to get trials atour site. The goal is to offer trial participation to our patients who otherwise might not be able to participate while offsetting some of the costs of immunotherapy. We are trying to make what could be a negative into a positive.
Measuring Success
Dr. Kambhampati. Unfortunately, we do not have any calculators to incorporate the quality of lives saved to the society. I know there are clearmetrics in transplant and in hematology, but unfortunately, there are no established metrics in solid tumor treatment that allow us to predict the cost savings to the health care system or to society or the benefit to the society. I don’t use any such predictive models or metrics in my decision making. These decisions are made based on existing evidence, and the existing evidence overwhelmingly supports use of immuno-oncology in certain types of solid tumors and in a select group of hematologic malignancies.
Dr. Kaster. This is where you can get more bang for your buck with an oncology pharmacist these days. A pharmacist can make a minor dosing change that will allow the same benefit for the patient, but could equal tens of thousands of dollars in cost-benefit for the VA. They can also be the second set of eyes when adjudicating a nonformulary request to ensure that a patient will benefit.
Dr. Lynch. Inappropriate prescribing is far more expensive than appropriate treatment. And the care for veterans whose long-term health outcomes could be improved by the new immunotherapies. It’s cheaper for veterans to be healthy and live longer than it is to take care of them in
their last 6 weeks of life. Unfortunately, there are not a lot of studies that have demonstrated that empirically, but I think it’s important to do those studies.
Role of Pharmacists
Dr. Lynch. I was at a meeting recently talking about how to improve veteran access to clinical trials. Francesca Cunningham, PharmD, director of the VA Center for Medication Safety of the VA Pharmacy Benefit Management Service (PBM) described the commitment that pharmacy has in taking a leadership role in the integration of precision medicine. Linking veterans’ tumor mutation status and pharmacogenetic variants to pharmacy databases is the best way to ensure treatment is informed by genetics. We have to be realistic about what we’re asking community oncologists to do. With the onset of precision oncology, 10 cancers have become really 100 cancers. In the prior model of care, it was the oncologist, maybe in collaboration with a pathologist, but it was mostly oncologists who determined care.
And in the evolution of precision oncology, Ithink that it’s become an interdisciplinary adventure. Pharmacy is going to play an increasinglyimportant role in precision medicine around all of the molecular alterations, even immuno-oncology regardless of molecular status in which the VA has an advantage. We’re not talking about some community pharmacist. We’re talking about a national health care system where there’s a national EHR, where there’s national PBM systems. So my thoughts on this aspect is that it’s an intricate multidisciplinary team who can ensure that veteran sget the best care possible: the best most cost-effective care possible.
Dr. Kaster. As an oncology pharmacist, I have to second that.
Ms. Nason. As Dr. Kaster said earlier, having a dedicated oncology pharmacist is tremendouslybeneficial. The oncology/hematology pharmacists are following the patients closely and notice when dose adjustments need to be made, optimizing the drug benefit and providing additional safety. Not to mention the cost benefit that can be realized with appropriate adjustment and the expertise they bring to managing possible interactionsand pharmacodynamics.
Dr. Kambhampati. To brag about the Kansas City VAMC program, we have published in Federal Practitioner our best practices showing the collaboration between a pharmacist and providers.6 And we have used several examples of cost savings, which have basically helped us build the research program, and several examples of dual monitoring oral chemotherapy monitoring. And we have created these templates within the EHR that allow everyone to get a quick snapshot of where things are, what needs to be done, and what needs to be monitored.
Now, we are taking it a step further to determine when to stop chemotherapy or when to stop treatments. For example, for chronic myeloid leukemia (CML), there are good data onstopping tyrosine kinase inhibitors.7 And that alone, if implemented across the VA, could bring
in huge cost savings, which perhaps could be put into investments in immuno-oncology or other efforts. We have several examples here that we have published, and we continue to increaseand strengthen our collaboration withour oncology pharmacist. We are very lucky and privileged to have a dedicated oncology pharmacistfor clinics and for research.
Dr. Lynch. The example of CML is perfect, because precision oncology has increased the complexity of care substantially. The VA is wellpositioned to be a leader in this area when care becomes this complex because of its ability to measure access to testing, to translate the results
of testing to pharmacy, to have pharmacists take the lead on prescribing, to have pathologists take the lead on molecular alterations, and to have oncologists take the lead on delivering the cancer care to the patients.
With hematologic malignancies, adherence in the early stages can result in patients getting offcare sooner, which is cost savings. But that requires access to testing, monitoring that testing, and working in partnership with pharmacy. This is a great story about how the VA is positioned to lead in this area of care.
Dr. Kaster. I would like to put a plug in for advanced practice providers and the use of nurse practitioners (NPs) and physician assistants (PAs).The VA is well positioned because it often has established interdisciplinary teams with these providers, pharmacy, nursing, and often social work, to coordinate the care and manage symptoms outside of oncologist visits.
Dr. Lynch. In the NCI cancer center model, once the patient has become stable, the ongoing careis designated to the NP or PA. Then as soon as there’s a change and it requires reevaluation, the oncologist becomes involved again. That pointabout the oncology treatment team is totally in line
with some of the previous comments.
Areas For Further Investigation
Dr. Kaster. There are so many nuances that we’re finding out all of the time about immunotherapies. A recent study brought up the role of antibiotics in the 30 or possibly 60 days prior to immunotherapy.3 How does that change treatment? Which patients are more likely to benefit from immunotherapies, and which are susceptible to “hyperprogression”? How do we integrate palliative care discussions into the carenow that patients are feeling better on treatment and may be less likely to want to discuss palliative care?
Ms. Nason. I absolutely agree with that, especially keeping palliative care integrated within our services. Our focus is now a little different, in thatwe have more optimistic outcomes in mind, butthere still are symptoms and issues where our colleaguesin palliative care are invaluable.
Dr. Lynch. I third that motion. What I would really like to see come out of this discussion is how veterans are getting access to leading oncology care. We just published an analysis of Medicare data and access to EGFR testing. The result of that analysis showed that testing in the VA was consistent with testing in Medicare.
For palliative care, I think the VA does a better job. And it’s just so discouraging as VA employees and as clinicians treating veterans to see publicationsthat suggest that veterans are getting a lower quality of care and that they would be better if care was privatized or outsourced. It’s just fundamentally not the case.
In CML, we see it. We’ve analyzed the data, in that there’s a far lower number of patients with CML who are included in the registry because patients who are diagnosed outside the VA are incorporated in other cancer registries.8 But as soon as their copays increase for access to targeted drugs, they immediately activate their VA benefits so that theycan get their drugs at the VA. For hematologic malignancies that are diagnosed outside the VA and are captured in other cancer registries, as soon as the drugs become expensive, they start getting their care in the VA. I don’t think there’s beena lot of empirical research that’s shown this, but we have the data to illustrate this trend. I hope thatthere are more publications that show that veterans with cancer are getting really good care inside the VA in the existing VA health care system.
Ms. Nason. It is disheartening to see negativepublicity, knowing that I work with colleagues who are strongly committed to providing up-to-date and relevant oncology care.
Dr. Lynch. As we record this conversation, I am in Rotterdam, Netherlands, in a meeting about genomewide testing. In hematologic malignancies, prostate cancer, and breast cancer, it’s a huge issue. And that is the other area that MVP (Million Veteran Program) is leading the way with the MVP biorepository data. Frankly, there’s no other biorepository that has this many patients, that has so many African Americans, and that has such rich EHR data. So inthat other area, the VA is doing really well.
1. Reck M, Rodríguez-Abreu D, Robinson AG, et al; KEYNOTE-024 Investigators. Pembrolizumab vs chemotherapy for PD-L1-positive non-small cell lung cancer. N Engl J Med. 2016;375(19):1823-1833.
2. Antonia SJ, Villegas A, Daniel D, et al; PACIFIC Investigators. Durvalumab after chemoradiotherapy in stage III non–smallcell lung cancer. N Engl J Med. 2017;377(20):1919-1929.
3. Hellmann MD, Ciuleanu T-E, Pluzansk A, et al. Nivolumab plus ipilimumab in Lung Cancer with a high tumor mutational burden. N Engl J Med. 2018 April 16. [Epub ahead of print.]
4. Motzer RJ, Tannir NM, McDermott DF, et al; CheckMate214 Investigators. Nivolumab plus ipilimumab versus sunitinibin advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277-1290.
5. Derosa L, Hellmann MD, Spaziano M, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small cell
lung cancer. Ann Oncol. 2018 March 30. [Epub ahead of print.]
6. Heinrichs A, Dessars B, El Housni H, et al. Identification of chronic myeloid leukemia patients treated with imatinib who are potentially eligible for treatment discontinuation by assessingreal-life molecular responses on the international scale in a EUTOS-certified lab. Leuk Res. 2018;67:27-31.
7. Keefe S, Kambhampati S, Powers B. An electronic chemotherapy ordering process and template. Fed Pract. 2015;32(suppl 1):21S-25S.
8. Lynch JA, Berse B, Rabb M, et al. Underutilization and disparities in access to EGFR testing among Medicare patients with lung cancer from 2010 - 2013. BMC Cancer. 2018;18(1):306.
The following is a lightly edited transcript of a teleconference discussion recorded in April 2018.
Suman Kambhampati, MD. Immuno-oncology is a paradigm-shifting treatment approach. It is an easy-to-understand term for both providers and for patients. The underlying principle is that the body’s own immune system is used or stimulated to fight cancer, and there are drugs that clearly have shown huge promise for this, not only in oncology, but also for other diseases. Time will tell whether that really pans out or not, but to begin with, the emphasis has been inoncology, and therefore, the term immunooncology is fitting.
Dr. Kaster. It was encouraging at first, especially when ipilimumab came out, to see the effects on patients with melanoma. Then the KEYNOTE-024 trial came out, and we were able to jump in anduse monoclonal antibodies directed against programmed death 1 (PD-1) in the first line, which is when things got exciting.1 We have a smaller populationin Boise, so PD-1s in lung cancer have had the biggest impact on our patients so far.
Ellen Nason, RN, MSN. Patients are open to immunotherapies.They’re excited about it. And as the other panelists have said, you can start broadly, as the body fights the cancer on its own, to providing more specific details as a patient wants more information. Immuno-oncology is definitely accepted by patients, and they’re very excited about it, especially with all the news about new therapies.
Dr. Kambhampati. For the Department of Veteran Affairs (VA) population, lung cancer has seen significant impact, and now it’s translating into other diseases through more research, trials, and better understanding about how these drugs are used and work.
The paradigm is shifting toward offering these drugs not only in metastatic cancers, but also in the surgically resectable tumors. The 2018 American Association for Cancer Research (AACR) meeting, just concluded. At the meeting several abstracts reported instances where immunooncology drugs are being introduced in the early phases of lung cancer and showing outstanding results. It’s very much possible that we’re going to see less use of traditional chemotherapy in the near future.
Ms. Nason. I primarily work with solid tumors,and the majority of the population I work with have lung cancer. So we’re excited about some of the results that we’ve seen and the lower toxicity involved. Recently, we’ve begun using durvalumab with patients with stage III disease. We have about 5 people now that are using it as a maintenance or consolidative treatment vs just using it for patients with stage IV disease. Hopefully, we’ll see some of the same results describedin the paper published on it.2
Dr. Kaster. Yes, we are incorporating these new changes into care as they're coming out. As Ms. Nason mentioned, we're already using immunotherapies in earlier settings, and we are seeing as much research that could be translated into care soon, like combining immunotherapies
in first-line settings, as we see in the Checkmate-227 study with nivolumab and ipilimumab.3,4 The landscape is going to change dramatically in the next couple of years.
Accessing Testing For First-Line Treatments
Dr. Lynch. There has been an ongoing discussionin the literature on accessing appropriate testing—delays in testing can result in patients who are not able to access the best targeted drugs on a first-line basis. The drug companiesand the VA have become highly sensitized to ensuring that veterans are accessing the appropriate testing. We are expanding the capability of VA labs to do that testing.
Ms. Nason. I want to put in a plug for the VA Precision Oncology Program (POP). It’s about 2 years into its existence, and Neil Spector, MD, is the director. The POP pays for sequencing the tumor samples.
A new sequencing contract will go into effect October 2018 and will include sequencing for hematologic malignancies in addition to the current testing of solid tumors. Patients from New York who have been unable to receive testing through the current vendors used by POP, will be included in the new contract. It is important to note that POP is working closely with the National Pharmacy Benefit Management Service (PBM) to develop a policy for approving off-label use of US Food and Drug Administration-approved targeted therapies based on sequenced data collected on patients tested through POP.
In addition, the leadership of POP is working to leverage the molecular testing results conducted through POP to improve veterans' access to clinical trials, both inside and outside the VA. Within the VA people can access information at tinyurl.com/precisiononcology. There is no reason why any eligible patient with cancer in the VA health care system should not have their tumor tissue sequenced through POP, particularly once the new contract goes into effect.
Dr. Lynch. Fortunately, the cost of next-generation sequencing has come down so much that most VA contracted reference laboratories offer next-generation sequencing, including LabCorp (Burlington,NC), Quest Diagnostics (Secaucus, NJ), Fulgent (Temple City, CA), and academic partners such as Oregon Health Sciences University and University of Washington.
Ms. Nason. At the Durham VAMC, sometimes a lack of tissue has been a barrier, but we now have the ability to send blood (liquid biopsy) for next-generation sequencing. Hopefully that will open up options for veterans with inadequate tissue. Importantly, all VA facilities can request liquid biopsiesthrough POP.
Dr. Lynch. That’s an important point. There have been huge advances in liquid biopsy testing.The VA Salt Lake City Health Care System (VASLCHCS) was in talks with Genomic Health (Redwood City, CA) to do a study as part of clinical operations to look at the concordance between the liquid biopsy testing and the precision oncology data. But Genomic Health eventually abandoned its liquid biopsy testing. Currently, the VA is only reimbursing or encouraging liquid biopsy if the tissue is not available or if the veteran has too high a level of comorbidities to undergo tissue biopsy. The main point for the discussion today is that access to testing is a key component of access to all of these advanced drugs.
Dr. Kambhampati. The precision medicine piece will be a game changer—no question about that. Liquid biopsy is very timely. Many patients have difficulty getting rebiopsied, so liquid biopsy is definitely a big, big step forward.
Still, there has not been consistency across the VA as there should be. Perhaps there are a few select centers, including our site in Kansas City, where access to precision medicine is readily available and liquid biopsies are available. We use the PlasmaSELECT test from Personal Genome Diagnostics (Baltimore, MD). We have just added Foundation Medicine (Cambridge, MA) also in hematology. Access to mutational profilingis absolutely a must for precision medicine.
All that being said, the unique issue with immuno-oncology is that it pretty much transcends the mutational profile and perhaps has leveled the playing field, irrespective of the tumor mutation profile or burden. In some solid tumors these immuno-oncology drugs have been shown to work across tumor types and across different mutation types. And there is a hint now in the recent data presented at AACR and in the New England Journalof Medicine showing that the tumor mutational burden is a predictor of pathologic response to at least PD-1 blockade in the resectable stages of lung cancer.1,3 To me, that’s a very important piece of data because that’s something that can be tested and can have a prognostic impact in immuno-oncology, particularly in the early stages of lung cancer and is further proof of the broad value of immunotherapics in targeting tumors irrespective of the precise tumor targets.
Dr. Kaster. Yes, it’s nice to see other options like tumor mutational burden and Lung Immune Prognostic Index being studied.5 It would be nice if we could rely a little more on these, and not PD-L1, which as we all know is a variable and an unreliable target.
Dr. Kambhampati. I agree.
Rural Challenges In A Veterans Population
Dr. Lynch. Providing high-quality cancer care to rural veterans care can be a challenge but it is a VA priority. The VA National Genomic Medicine Services offers better access for rural veterans to germline genetic testing than any other healthcare system in the country. In terms of access to somatic testing and next-generation sequencing, we are working toward providing the same level of cancer care as patients would receive at National Cancer Institute (NCI) cancer centers. The VA oncology leadership has done teleconsults and virtual tumor boards, but for some rural VAMCs, fellowsare leading the clinical care. As we expand use of oral agents for oncology treatment, it will be easier to ensure that rural veterans receive the same standard of care for POP that veterans being cared for at VASLCHCS, Kansas City VAMC, or Durham VAMC get.
Dr. Kambhampati. The Kansas City VAMC in its catchment area includes underserved areas, such as Topeka and Leavenworth, Kansas. What we’ve been able to do here is something that’s unique—Kansas City VAMC is the only standalone VA in the country to be recognized as a primary SWOG (Southwestern Oncology Group) institution, which provides access to many trials, such as the Lung-MAP trial and others. And that has allowed us to use the full expanse of precision medicine without financial barriers. The research has helped us improve the standard of
care for patients across VISN 15.
Dr. Lynch. In precision oncology, the chief of pathology is an important figure in access to advanced care. I’ve worked with Sharad Mathur,MD, of the Kansas City VAMC on many clinical trials. He’s on the Kansas City VAMC Institutional Review Board and the cancer committee and is tuned in to veterans’ access to precision oncology. Kansas City was ordering Foundation One for select patients that met the criteria probably sooner than any other VA and participated in NCI Cooperative Group clinical trials. It is a great example of how veterans are getting access to
the same level of care as are patients who gettreated at NCI partners.
Comorbidities
Dr. Kambhampati. I don’t treat a lot of patients with lung cancer, but I find it easier to use these immuno-oncology drugs than platinums and etoposide. I consider them absolutely nasty chemotherapy drugs now in this era of immuno-oncology and targeted therapy.
Dr. Lynch. The VA is very important in translational lung cancer research and clinical care. It used to be thought that African American patients don’t get epidermal growth factor receptor mutations. And that’s because not enough African American patients with lung cancer were included in the NCI-based clinical trial.There are7,000 veterans who get lung cancer each year, and 20% to 25% of those are African Americans. Prevalence of various mutations and the pharmacogenetics of some of these drugs differ by patient ancestry. Including veterans with lung
cancer in precision oncology clinical trials and clinical care is not just a priority for the VA but a priority for NCI and internationally. I can’t emphasize this enough—veterans with lung cancer should be included in these studies and should be getting the same level of care that our partners are getting at NCI cancer centers. In the VA we’re positioned to do this because of our nationalelectronic health record (EHR) and becauseof our ability to identify patients with specific variants and enroll them in clinical trials.
Ms. Nason. One of the barriers that I find withsome of the patients that I have treated is getting them to a trial. If the trial isn’t available locally, specifically there are socioeconomic and distance issues that are hard to overcome.
Dr. Kaster. For smaller medical centers, getting patients to clinical trials can be difficult. The Boise VAMC is putting together a proposal now to justify hiring a research pharmacist in order to get trials atour site. The goal is to offer trial participation to our patients who otherwise might not be able to participate while offsetting some of the costs of immunotherapy. We are trying to make what could be a negative into a positive.
Measuring Success
Dr. Kambhampati. Unfortunately, we do not have any calculators to incorporate the quality of lives saved to the society. I know there are clearmetrics in transplant and in hematology, but unfortunately, there are no established metrics in solid tumor treatment that allow us to predict the cost savings to the health care system or to society or the benefit to the society. I don’t use any such predictive models or metrics in my decision making. These decisions are made based on existing evidence, and the existing evidence overwhelmingly supports use of immuno-oncology in certain types of solid tumors and in a select group of hematologic malignancies.
Dr. Kaster. This is where you can get more bang for your buck with an oncology pharmacist these days. A pharmacist can make a minor dosing change that will allow the same benefit for the patient, but could equal tens of thousands of dollars in cost-benefit for the VA. They can also be the second set of eyes when adjudicating a nonformulary request to ensure that a patient will benefit.
Dr. Lynch. Inappropriate prescribing is far more expensive than appropriate treatment. And the care for veterans whose long-term health outcomes could be improved by the new immunotherapies. It’s cheaper for veterans to be healthy and live longer than it is to take care of them in
their last 6 weeks of life. Unfortunately, there are not a lot of studies that have demonstrated that empirically, but I think it’s important to do those studies.
Role of Pharmacists
Dr. Lynch. I was at a meeting recently talking about how to improve veteran access to clinical trials. Francesca Cunningham, PharmD, director of the VA Center for Medication Safety of the VA Pharmacy Benefit Management Service (PBM) described the commitment that pharmacy has in taking a leadership role in the integration of precision medicine. Linking veterans’ tumor mutation status and pharmacogenetic variants to pharmacy databases is the best way to ensure treatment is informed by genetics. We have to be realistic about what we’re asking community oncologists to do. With the onset of precision oncology, 10 cancers have become really 100 cancers. In the prior model of care, it was the oncologist, maybe in collaboration with a pathologist, but it was mostly oncologists who determined care.
And in the evolution of precision oncology, Ithink that it’s become an interdisciplinary adventure. Pharmacy is going to play an increasinglyimportant role in precision medicine around all of the molecular alterations, even immuno-oncology regardless of molecular status in which the VA has an advantage. We’re not talking about some community pharmacist. We’re talking about a national health care system where there’s a national EHR, where there’s national PBM systems. So my thoughts on this aspect is that it’s an intricate multidisciplinary team who can ensure that veteran sget the best care possible: the best most cost-effective care possible.
Dr. Kaster. As an oncology pharmacist, I have to second that.
Ms. Nason. As Dr. Kaster said earlier, having a dedicated oncology pharmacist is tremendouslybeneficial. The oncology/hematology pharmacists are following the patients closely and notice when dose adjustments need to be made, optimizing the drug benefit and providing additional safety. Not to mention the cost benefit that can be realized with appropriate adjustment and the expertise they bring to managing possible interactionsand pharmacodynamics.
Dr. Kambhampati. To brag about the Kansas City VAMC program, we have published in Federal Practitioner our best practices showing the collaboration between a pharmacist and providers.6 And we have used several examples of cost savings, which have basically helped us build the research program, and several examples of dual monitoring oral chemotherapy monitoring. And we have created these templates within the EHR that allow everyone to get a quick snapshot of where things are, what needs to be done, and what needs to be monitored.
Now, we are taking it a step further to determine when to stop chemotherapy or when to stop treatments. For example, for chronic myeloid leukemia (CML), there are good data onstopping tyrosine kinase inhibitors.7 And that alone, if implemented across the VA, could bring
in huge cost savings, which perhaps could be put into investments in immuno-oncology or other efforts. We have several examples here that we have published, and we continue to increaseand strengthen our collaboration withour oncology pharmacist. We are very lucky and privileged to have a dedicated oncology pharmacistfor clinics and for research.
Dr. Lynch. The example of CML is perfect, because precision oncology has increased the complexity of care substantially. The VA is wellpositioned to be a leader in this area when care becomes this complex because of its ability to measure access to testing, to translate the results
of testing to pharmacy, to have pharmacists take the lead on prescribing, to have pathologists take the lead on molecular alterations, and to have oncologists take the lead on delivering the cancer care to the patients.
With hematologic malignancies, adherence in the early stages can result in patients getting offcare sooner, which is cost savings. But that requires access to testing, monitoring that testing, and working in partnership with pharmacy. This is a great story about how the VA is positioned to lead in this area of care.
Dr. Kaster. I would like to put a plug in for advanced practice providers and the use of nurse practitioners (NPs) and physician assistants (PAs).The VA is well positioned because it often has established interdisciplinary teams with these providers, pharmacy, nursing, and often social work, to coordinate the care and manage symptoms outside of oncologist visits.
Dr. Lynch. In the NCI cancer center model, once the patient has become stable, the ongoing careis designated to the NP or PA. Then as soon as there’s a change and it requires reevaluation, the oncologist becomes involved again. That pointabout the oncology treatment team is totally in line
with some of the previous comments.
Areas For Further Investigation
Dr. Kaster. There are so many nuances that we’re finding out all of the time about immunotherapies. A recent study brought up the role of antibiotics in the 30 or possibly 60 days prior to immunotherapy.3 How does that change treatment? Which patients are more likely to benefit from immunotherapies, and which are susceptible to “hyperprogression”? How do we integrate palliative care discussions into the carenow that patients are feeling better on treatment and may be less likely to want to discuss palliative care?
Ms. Nason. I absolutely agree with that, especially keeping palliative care integrated within our services. Our focus is now a little different, in thatwe have more optimistic outcomes in mind, butthere still are symptoms and issues where our colleaguesin palliative care are invaluable.
Dr. Lynch. I third that motion. What I would really like to see come out of this discussion is how veterans are getting access to leading oncology care. We just published an analysis of Medicare data and access to EGFR testing. The result of that analysis showed that testing in the VA was consistent with testing in Medicare.
For palliative care, I think the VA does a better job. And it’s just so discouraging as VA employees and as clinicians treating veterans to see publicationsthat suggest that veterans are getting a lower quality of care and that they would be better if care was privatized or outsourced. It’s just fundamentally not the case.
In CML, we see it. We’ve analyzed the data, in that there’s a far lower number of patients with CML who are included in the registry because patients who are diagnosed outside the VA are incorporated in other cancer registries.8 But as soon as their copays increase for access to targeted drugs, they immediately activate their VA benefits so that theycan get their drugs at the VA. For hematologic malignancies that are diagnosed outside the VA and are captured in other cancer registries, as soon as the drugs become expensive, they start getting their care in the VA. I don’t think there’s beena lot of empirical research that’s shown this, but we have the data to illustrate this trend. I hope thatthere are more publications that show that veterans with cancer are getting really good care inside the VA in the existing VA health care system.
Ms. Nason. It is disheartening to see negativepublicity, knowing that I work with colleagues who are strongly committed to providing up-to-date and relevant oncology care.
Dr. Lynch. As we record this conversation, I am in Rotterdam, Netherlands, in a meeting about genomewide testing. In hematologic malignancies, prostate cancer, and breast cancer, it’s a huge issue. And that is the other area that MVP (Million Veteran Program) is leading the way with the MVP biorepository data. Frankly, there’s no other biorepository that has this many patients, that has so many African Americans, and that has such rich EHR data. So inthat other area, the VA is doing really well.
The following is a lightly edited transcript of a teleconference discussion recorded in April 2018.
Suman Kambhampati, MD. Immuno-oncology is a paradigm-shifting treatment approach. It is an easy-to-understand term for both providers and for patients. The underlying principle is that the body’s own immune system is used or stimulated to fight cancer, and there are drugs that clearly have shown huge promise for this, not only in oncology, but also for other diseases. Time will tell whether that really pans out or not, but to begin with, the emphasis has been inoncology, and therefore, the term immunooncology is fitting.
Dr. Kaster. It was encouraging at first, especially when ipilimumab came out, to see the effects on patients with melanoma. Then the KEYNOTE-024 trial came out, and we were able to jump in anduse monoclonal antibodies directed against programmed death 1 (PD-1) in the first line, which is when things got exciting.1 We have a smaller populationin Boise, so PD-1s in lung cancer have had the biggest impact on our patients so far.
Ellen Nason, RN, MSN. Patients are open to immunotherapies.They’re excited about it. And as the other panelists have said, you can start broadly, as the body fights the cancer on its own, to providing more specific details as a patient wants more information. Immuno-oncology is definitely accepted by patients, and they’re very excited about it, especially with all the news about new therapies.
Dr. Kambhampati. For the Department of Veteran Affairs (VA) population, lung cancer has seen significant impact, and now it’s translating into other diseases through more research, trials, and better understanding about how these drugs are used and work.
The paradigm is shifting toward offering these drugs not only in metastatic cancers, but also in the surgically resectable tumors. The 2018 American Association for Cancer Research (AACR) meeting, just concluded. At the meeting several abstracts reported instances where immunooncology drugs are being introduced in the early phases of lung cancer and showing outstanding results. It’s very much possible that we’re going to see less use of traditional chemotherapy in the near future.
Ms. Nason. I primarily work with solid tumors,and the majority of the population I work with have lung cancer. So we’re excited about some of the results that we’ve seen and the lower toxicity involved. Recently, we’ve begun using durvalumab with patients with stage III disease. We have about 5 people now that are using it as a maintenance or consolidative treatment vs just using it for patients with stage IV disease. Hopefully, we’ll see some of the same results describedin the paper published on it.2
Dr. Kaster. Yes, we are incorporating these new changes into care as they're coming out. As Ms. Nason mentioned, we're already using immunotherapies in earlier settings, and we are seeing as much research that could be translated into care soon, like combining immunotherapies
in first-line settings, as we see in the Checkmate-227 study with nivolumab and ipilimumab.3,4 The landscape is going to change dramatically in the next couple of years.
Accessing Testing For First-Line Treatments
Dr. Lynch. There has been an ongoing discussionin the literature on accessing appropriate testing—delays in testing can result in patients who are not able to access the best targeted drugs on a first-line basis. The drug companiesand the VA have become highly sensitized to ensuring that veterans are accessing the appropriate testing. We are expanding the capability of VA labs to do that testing.
Ms. Nason. I want to put in a plug for the VA Precision Oncology Program (POP). It’s about 2 years into its existence, and Neil Spector, MD, is the director. The POP pays for sequencing the tumor samples.
A new sequencing contract will go into effect October 2018 and will include sequencing for hematologic malignancies in addition to the current testing of solid tumors. Patients from New York who have been unable to receive testing through the current vendors used by POP, will be included in the new contract. It is important to note that POP is working closely with the National Pharmacy Benefit Management Service (PBM) to develop a policy for approving off-label use of US Food and Drug Administration-approved targeted therapies based on sequenced data collected on patients tested through POP.
In addition, the leadership of POP is working to leverage the molecular testing results conducted through POP to improve veterans' access to clinical trials, both inside and outside the VA. Within the VA people can access information at tinyurl.com/precisiononcology. There is no reason why any eligible patient with cancer in the VA health care system should not have their tumor tissue sequenced through POP, particularly once the new contract goes into effect.
Dr. Lynch. Fortunately, the cost of next-generation sequencing has come down so much that most VA contracted reference laboratories offer next-generation sequencing, including LabCorp (Burlington,NC), Quest Diagnostics (Secaucus, NJ), Fulgent (Temple City, CA), and academic partners such as Oregon Health Sciences University and University of Washington.
Ms. Nason. At the Durham VAMC, sometimes a lack of tissue has been a barrier, but we now have the ability to send blood (liquid biopsy) for next-generation sequencing. Hopefully that will open up options for veterans with inadequate tissue. Importantly, all VA facilities can request liquid biopsiesthrough POP.
Dr. Lynch. That’s an important point. There have been huge advances in liquid biopsy testing.The VA Salt Lake City Health Care System (VASLCHCS) was in talks with Genomic Health (Redwood City, CA) to do a study as part of clinical operations to look at the concordance between the liquid biopsy testing and the precision oncology data. But Genomic Health eventually abandoned its liquid biopsy testing. Currently, the VA is only reimbursing or encouraging liquid biopsy if the tissue is not available or if the veteran has too high a level of comorbidities to undergo tissue biopsy. The main point for the discussion today is that access to testing is a key component of access to all of these advanced drugs.
Dr. Kambhampati. The precision medicine piece will be a game changer—no question about that. Liquid biopsy is very timely. Many patients have difficulty getting rebiopsied, so liquid biopsy is definitely a big, big step forward.
Still, there has not been consistency across the VA as there should be. Perhaps there are a few select centers, including our site in Kansas City, where access to precision medicine is readily available and liquid biopsies are available. We use the PlasmaSELECT test from Personal Genome Diagnostics (Baltimore, MD). We have just added Foundation Medicine (Cambridge, MA) also in hematology. Access to mutational profilingis absolutely a must for precision medicine.
All that being said, the unique issue with immuno-oncology is that it pretty much transcends the mutational profile and perhaps has leveled the playing field, irrespective of the tumor mutation profile or burden. In some solid tumors these immuno-oncology drugs have been shown to work across tumor types and across different mutation types. And there is a hint now in the recent data presented at AACR and in the New England Journalof Medicine showing that the tumor mutational burden is a predictor of pathologic response to at least PD-1 blockade in the resectable stages of lung cancer.1,3 To me, that’s a very important piece of data because that’s something that can be tested and can have a prognostic impact in immuno-oncology, particularly in the early stages of lung cancer and is further proof of the broad value of immunotherapics in targeting tumors irrespective of the precise tumor targets.
Dr. Kaster. Yes, it’s nice to see other options like tumor mutational burden and Lung Immune Prognostic Index being studied.5 It would be nice if we could rely a little more on these, and not PD-L1, which as we all know is a variable and an unreliable target.
Dr. Kambhampati. I agree.
Rural Challenges In A Veterans Population
Dr. Lynch. Providing high-quality cancer care to rural veterans care can be a challenge but it is a VA priority. The VA National Genomic Medicine Services offers better access for rural veterans to germline genetic testing than any other healthcare system in the country. In terms of access to somatic testing and next-generation sequencing, we are working toward providing the same level of cancer care as patients would receive at National Cancer Institute (NCI) cancer centers. The VA oncology leadership has done teleconsults and virtual tumor boards, but for some rural VAMCs, fellowsare leading the clinical care. As we expand use of oral agents for oncology treatment, it will be easier to ensure that rural veterans receive the same standard of care for POP that veterans being cared for at VASLCHCS, Kansas City VAMC, or Durham VAMC get.
Dr. Kambhampati. The Kansas City VAMC in its catchment area includes underserved areas, such as Topeka and Leavenworth, Kansas. What we’ve been able to do here is something that’s unique—Kansas City VAMC is the only standalone VA in the country to be recognized as a primary SWOG (Southwestern Oncology Group) institution, which provides access to many trials, such as the Lung-MAP trial and others. And that has allowed us to use the full expanse of precision medicine without financial barriers. The research has helped us improve the standard of
care for patients across VISN 15.
Dr. Lynch. In precision oncology, the chief of pathology is an important figure in access to advanced care. I’ve worked with Sharad Mathur,MD, of the Kansas City VAMC on many clinical trials. He’s on the Kansas City VAMC Institutional Review Board and the cancer committee and is tuned in to veterans’ access to precision oncology. Kansas City was ordering Foundation One for select patients that met the criteria probably sooner than any other VA and participated in NCI Cooperative Group clinical trials. It is a great example of how veterans are getting access to
the same level of care as are patients who gettreated at NCI partners.
Comorbidities
Dr. Kambhampati. I don’t treat a lot of patients with lung cancer, but I find it easier to use these immuno-oncology drugs than platinums and etoposide. I consider them absolutely nasty chemotherapy drugs now in this era of immuno-oncology and targeted therapy.
Dr. Lynch. The VA is very important in translational lung cancer research and clinical care. It used to be thought that African American patients don’t get epidermal growth factor receptor mutations. And that’s because not enough African American patients with lung cancer were included in the NCI-based clinical trial.There are7,000 veterans who get lung cancer each year, and 20% to 25% of those are African Americans. Prevalence of various mutations and the pharmacogenetics of some of these drugs differ by patient ancestry. Including veterans with lung
cancer in precision oncology clinical trials and clinical care is not just a priority for the VA but a priority for NCI and internationally. I can’t emphasize this enough—veterans with lung cancer should be included in these studies and should be getting the same level of care that our partners are getting at NCI cancer centers. In the VA we’re positioned to do this because of our nationalelectronic health record (EHR) and becauseof our ability to identify patients with specific variants and enroll them in clinical trials.
Ms. Nason. One of the barriers that I find withsome of the patients that I have treated is getting them to a trial. If the trial isn’t available locally, specifically there are socioeconomic and distance issues that are hard to overcome.
Dr. Kaster. For smaller medical centers, getting patients to clinical trials can be difficult. The Boise VAMC is putting together a proposal now to justify hiring a research pharmacist in order to get trials atour site. The goal is to offer trial participation to our patients who otherwise might not be able to participate while offsetting some of the costs of immunotherapy. We are trying to make what could be a negative into a positive.
Measuring Success
Dr. Kambhampati. Unfortunately, we do not have any calculators to incorporate the quality of lives saved to the society. I know there are clearmetrics in transplant and in hematology, but unfortunately, there are no established metrics in solid tumor treatment that allow us to predict the cost savings to the health care system or to society or the benefit to the society. I don’t use any such predictive models or metrics in my decision making. These decisions are made based on existing evidence, and the existing evidence overwhelmingly supports use of immuno-oncology in certain types of solid tumors and in a select group of hematologic malignancies.
Dr. Kaster. This is where you can get more bang for your buck with an oncology pharmacist these days. A pharmacist can make a minor dosing change that will allow the same benefit for the patient, but could equal tens of thousands of dollars in cost-benefit for the VA. They can also be the second set of eyes when adjudicating a nonformulary request to ensure that a patient will benefit.
Dr. Lynch. Inappropriate prescribing is far more expensive than appropriate treatment. And the care for veterans whose long-term health outcomes could be improved by the new immunotherapies. It’s cheaper for veterans to be healthy and live longer than it is to take care of them in
their last 6 weeks of life. Unfortunately, there are not a lot of studies that have demonstrated that empirically, but I think it’s important to do those studies.
Role of Pharmacists
Dr. Lynch. I was at a meeting recently talking about how to improve veteran access to clinical trials. Francesca Cunningham, PharmD, director of the VA Center for Medication Safety of the VA Pharmacy Benefit Management Service (PBM) described the commitment that pharmacy has in taking a leadership role in the integration of precision medicine. Linking veterans’ tumor mutation status and pharmacogenetic variants to pharmacy databases is the best way to ensure treatment is informed by genetics. We have to be realistic about what we’re asking community oncologists to do. With the onset of precision oncology, 10 cancers have become really 100 cancers. In the prior model of care, it was the oncologist, maybe in collaboration with a pathologist, but it was mostly oncologists who determined care.
And in the evolution of precision oncology, Ithink that it’s become an interdisciplinary adventure. Pharmacy is going to play an increasinglyimportant role in precision medicine around all of the molecular alterations, even immuno-oncology regardless of molecular status in which the VA has an advantage. We’re not talking about some community pharmacist. We’re talking about a national health care system where there’s a national EHR, where there’s national PBM systems. So my thoughts on this aspect is that it’s an intricate multidisciplinary team who can ensure that veteran sget the best care possible: the best most cost-effective care possible.
Dr. Kaster. As an oncology pharmacist, I have to second that.
Ms. Nason. As Dr. Kaster said earlier, having a dedicated oncology pharmacist is tremendouslybeneficial. The oncology/hematology pharmacists are following the patients closely and notice when dose adjustments need to be made, optimizing the drug benefit and providing additional safety. Not to mention the cost benefit that can be realized with appropriate adjustment and the expertise they bring to managing possible interactionsand pharmacodynamics.
Dr. Kambhampati. To brag about the Kansas City VAMC program, we have published in Federal Practitioner our best practices showing the collaboration between a pharmacist and providers.6 And we have used several examples of cost savings, which have basically helped us build the research program, and several examples of dual monitoring oral chemotherapy monitoring. And we have created these templates within the EHR that allow everyone to get a quick snapshot of where things are, what needs to be done, and what needs to be monitored.
Now, we are taking it a step further to determine when to stop chemotherapy or when to stop treatments. For example, for chronic myeloid leukemia (CML), there are good data onstopping tyrosine kinase inhibitors.7 And that alone, if implemented across the VA, could bring
in huge cost savings, which perhaps could be put into investments in immuno-oncology or other efforts. We have several examples here that we have published, and we continue to increaseand strengthen our collaboration withour oncology pharmacist. We are very lucky and privileged to have a dedicated oncology pharmacistfor clinics and for research.
Dr. Lynch. The example of CML is perfect, because precision oncology has increased the complexity of care substantially. The VA is wellpositioned to be a leader in this area when care becomes this complex because of its ability to measure access to testing, to translate the results
of testing to pharmacy, to have pharmacists take the lead on prescribing, to have pathologists take the lead on molecular alterations, and to have oncologists take the lead on delivering the cancer care to the patients.
With hematologic malignancies, adherence in the early stages can result in patients getting offcare sooner, which is cost savings. But that requires access to testing, monitoring that testing, and working in partnership with pharmacy. This is a great story about how the VA is positioned to lead in this area of care.
Dr. Kaster. I would like to put a plug in for advanced practice providers and the use of nurse practitioners (NPs) and physician assistants (PAs).The VA is well positioned because it often has established interdisciplinary teams with these providers, pharmacy, nursing, and often social work, to coordinate the care and manage symptoms outside of oncologist visits.
Dr. Lynch. In the NCI cancer center model, once the patient has become stable, the ongoing careis designated to the NP or PA. Then as soon as there’s a change and it requires reevaluation, the oncologist becomes involved again. That pointabout the oncology treatment team is totally in line
with some of the previous comments.
Areas For Further Investigation
Dr. Kaster. There are so many nuances that we’re finding out all of the time about immunotherapies. A recent study brought up the role of antibiotics in the 30 or possibly 60 days prior to immunotherapy.3 How does that change treatment? Which patients are more likely to benefit from immunotherapies, and which are susceptible to “hyperprogression”? How do we integrate palliative care discussions into the carenow that patients are feeling better on treatment and may be less likely to want to discuss palliative care?
Ms. Nason. I absolutely agree with that, especially keeping palliative care integrated within our services. Our focus is now a little different, in thatwe have more optimistic outcomes in mind, butthere still are symptoms and issues where our colleaguesin palliative care are invaluable.
Dr. Lynch. I third that motion. What I would really like to see come out of this discussion is how veterans are getting access to leading oncology care. We just published an analysis of Medicare data and access to EGFR testing. The result of that analysis showed that testing in the VA was consistent with testing in Medicare.
For palliative care, I think the VA does a better job. And it’s just so discouraging as VA employees and as clinicians treating veterans to see publicationsthat suggest that veterans are getting a lower quality of care and that they would be better if care was privatized or outsourced. It’s just fundamentally not the case.
In CML, we see it. We’ve analyzed the data, in that there’s a far lower number of patients with CML who are included in the registry because patients who are diagnosed outside the VA are incorporated in other cancer registries.8 But as soon as their copays increase for access to targeted drugs, they immediately activate their VA benefits so that theycan get their drugs at the VA. For hematologic malignancies that are diagnosed outside the VA and are captured in other cancer registries, as soon as the drugs become expensive, they start getting their care in the VA. I don’t think there’s beena lot of empirical research that’s shown this, but we have the data to illustrate this trend. I hope thatthere are more publications that show that veterans with cancer are getting really good care inside the VA in the existing VA health care system.
Ms. Nason. It is disheartening to see negativepublicity, knowing that I work with colleagues who are strongly committed to providing up-to-date and relevant oncology care.
Dr. Lynch. As we record this conversation, I am in Rotterdam, Netherlands, in a meeting about genomewide testing. In hematologic malignancies, prostate cancer, and breast cancer, it’s a huge issue. And that is the other area that MVP (Million Veteran Program) is leading the way with the MVP biorepository data. Frankly, there’s no other biorepository that has this many patients, that has so many African Americans, and that has such rich EHR data. So inthat other area, the VA is doing really well.
1. Reck M, Rodríguez-Abreu D, Robinson AG, et al; KEYNOTE-024 Investigators. Pembrolizumab vs chemotherapy for PD-L1-positive non-small cell lung cancer. N Engl J Med. 2016;375(19):1823-1833.
2. Antonia SJ, Villegas A, Daniel D, et al; PACIFIC Investigators. Durvalumab after chemoradiotherapy in stage III non–smallcell lung cancer. N Engl J Med. 2017;377(20):1919-1929.
3. Hellmann MD, Ciuleanu T-E, Pluzansk A, et al. Nivolumab plus ipilimumab in Lung Cancer with a high tumor mutational burden. N Engl J Med. 2018 April 16. [Epub ahead of print.]
4. Motzer RJ, Tannir NM, McDermott DF, et al; CheckMate214 Investigators. Nivolumab plus ipilimumab versus sunitinibin advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277-1290.
5. Derosa L, Hellmann MD, Spaziano M, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small cell
lung cancer. Ann Oncol. 2018 March 30. [Epub ahead of print.]
6. Heinrichs A, Dessars B, El Housni H, et al. Identification of chronic myeloid leukemia patients treated with imatinib who are potentially eligible for treatment discontinuation by assessingreal-life molecular responses on the international scale in a EUTOS-certified lab. Leuk Res. 2018;67:27-31.
7. Keefe S, Kambhampati S, Powers B. An electronic chemotherapy ordering process and template. Fed Pract. 2015;32(suppl 1):21S-25S.
8. Lynch JA, Berse B, Rabb M, et al. Underutilization and disparities in access to EGFR testing among Medicare patients with lung cancer from 2010 - 2013. BMC Cancer. 2018;18(1):306.
1. Reck M, Rodríguez-Abreu D, Robinson AG, et al; KEYNOTE-024 Investigators. Pembrolizumab vs chemotherapy for PD-L1-positive non-small cell lung cancer. N Engl J Med. 2016;375(19):1823-1833.
2. Antonia SJ, Villegas A, Daniel D, et al; PACIFIC Investigators. Durvalumab after chemoradiotherapy in stage III non–smallcell lung cancer. N Engl J Med. 2017;377(20):1919-1929.
3. Hellmann MD, Ciuleanu T-E, Pluzansk A, et al. Nivolumab plus ipilimumab in Lung Cancer with a high tumor mutational burden. N Engl J Med. 2018 April 16. [Epub ahead of print.]
4. Motzer RJ, Tannir NM, McDermott DF, et al; CheckMate214 Investigators. Nivolumab plus ipilimumab versus sunitinibin advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277-1290.
5. Derosa L, Hellmann MD, Spaziano M, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small cell
lung cancer. Ann Oncol. 2018 March 30. [Epub ahead of print.]
6. Heinrichs A, Dessars B, El Housni H, et al. Identification of chronic myeloid leukemia patients treated with imatinib who are potentially eligible for treatment discontinuation by assessingreal-life molecular responses on the international scale in a EUTOS-certified lab. Leuk Res. 2018;67:27-31.
7. Keefe S, Kambhampati S, Powers B. An electronic chemotherapy ordering process and template. Fed Pract. 2015;32(suppl 1):21S-25S.
8. Lynch JA, Berse B, Rabb M, et al. Underutilization and disparities in access to EGFR testing among Medicare patients with lung cancer from 2010 - 2013. BMC Cancer. 2018;18(1):306.
Emerging biosimilars market presents opportunities and challenges
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6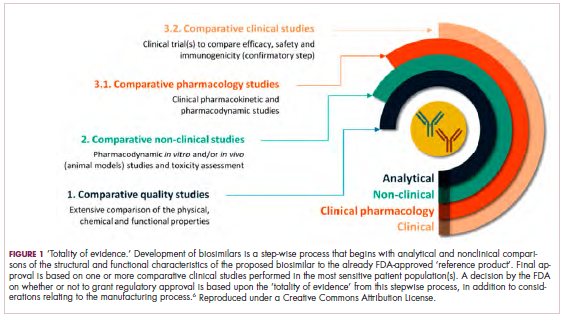
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10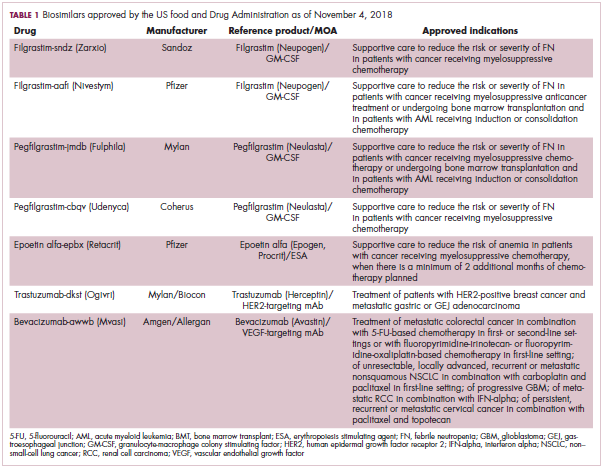
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22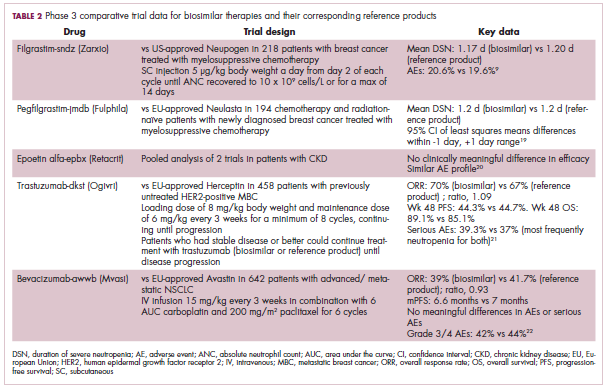
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.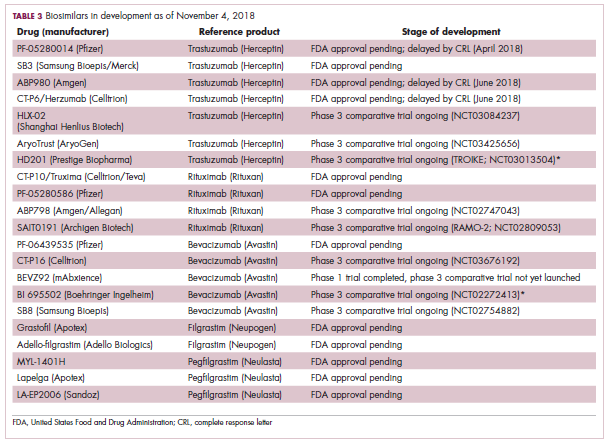
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
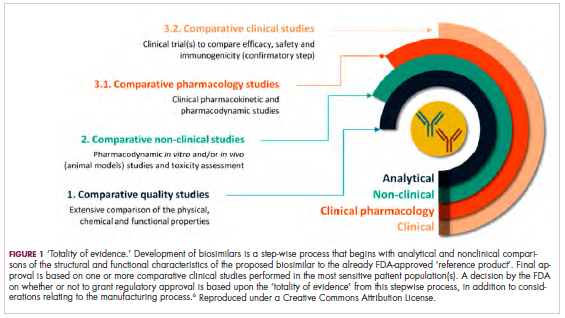
Immunotherapy may hold the key to defeating virally associated cancers
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.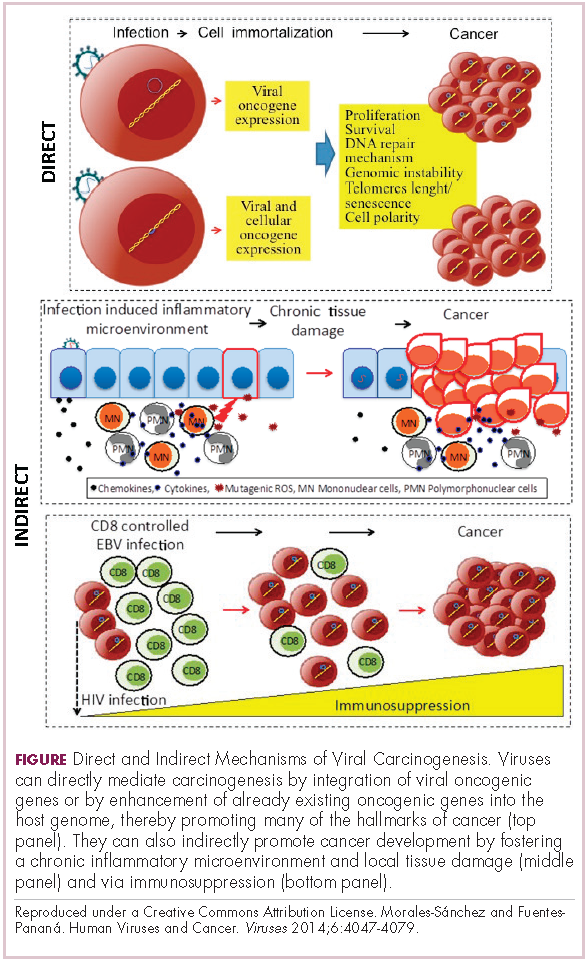
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).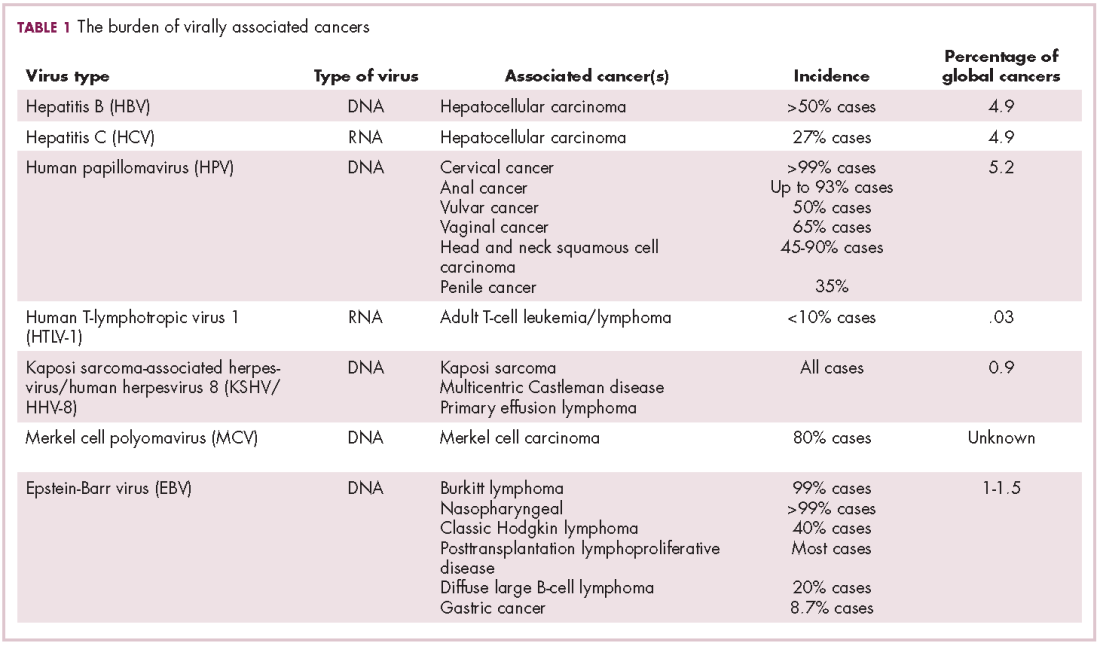
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9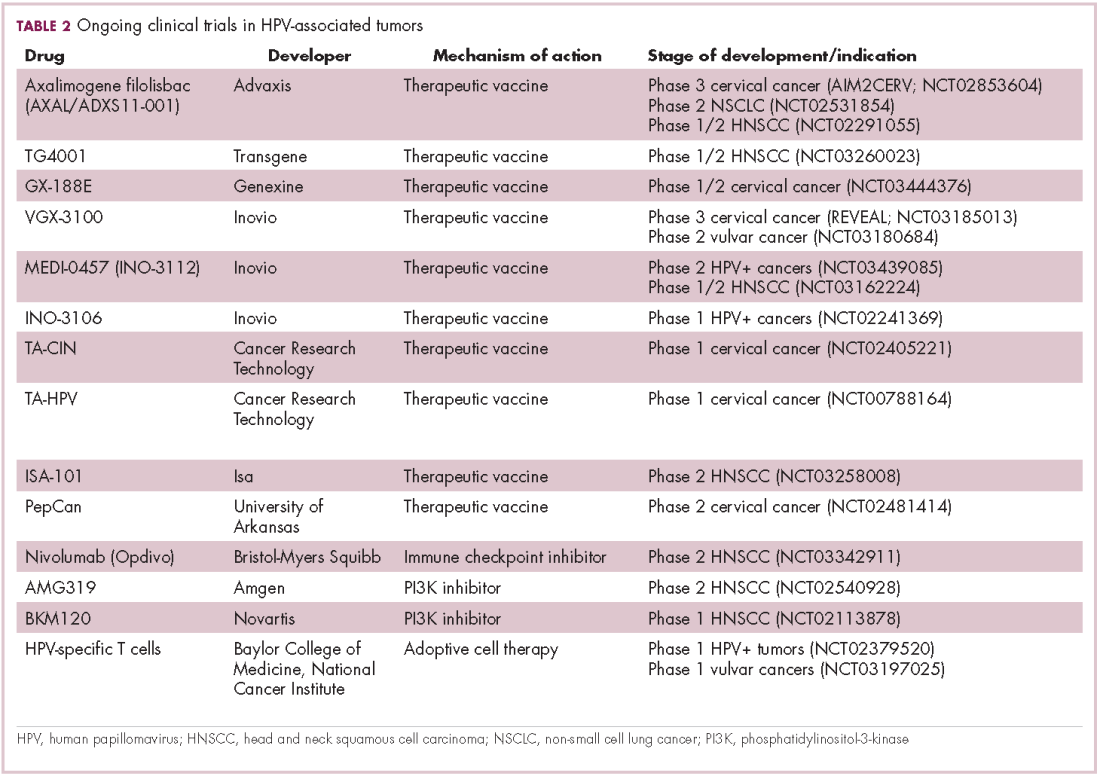
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).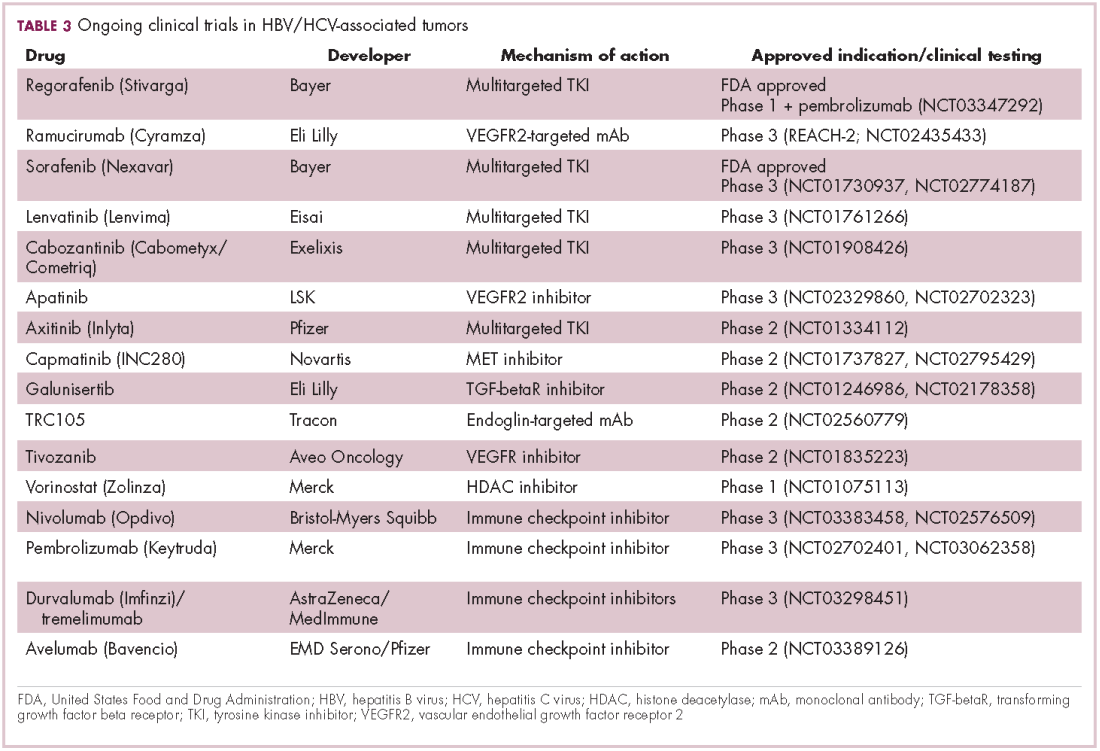
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).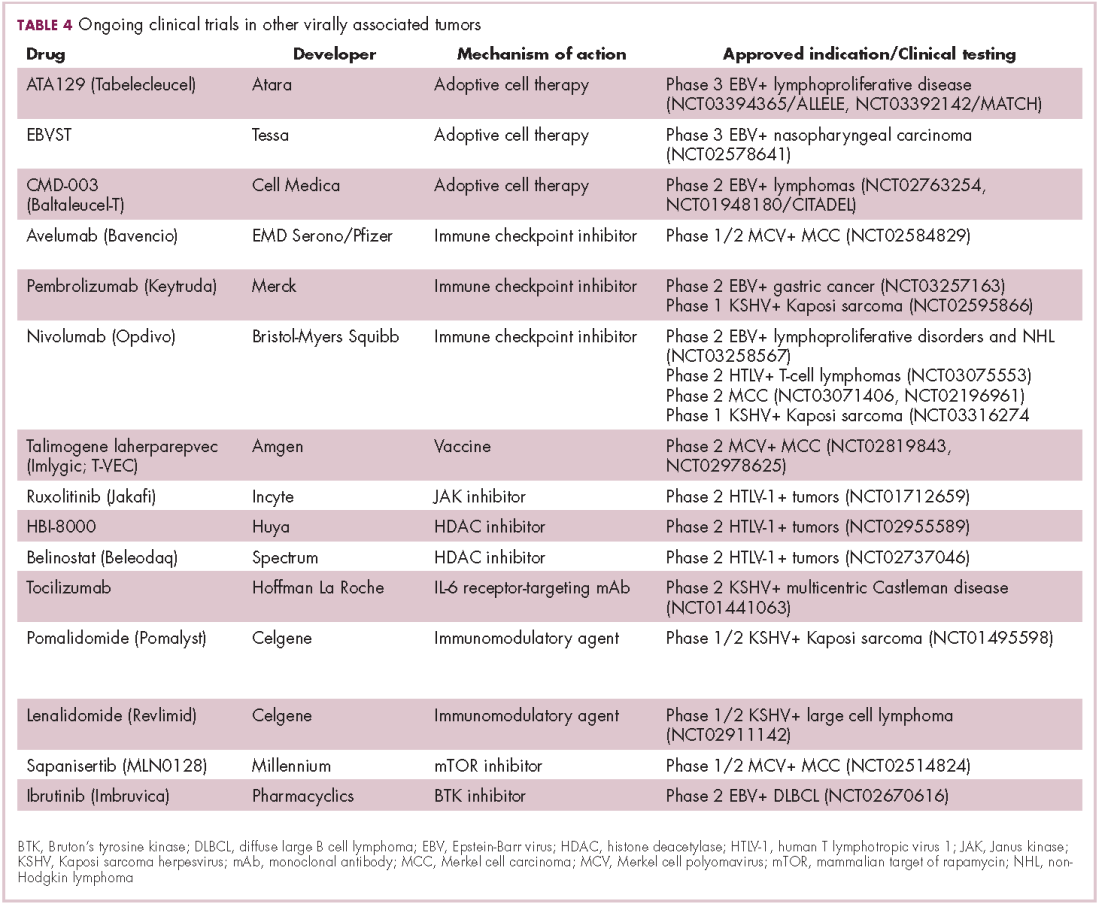
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
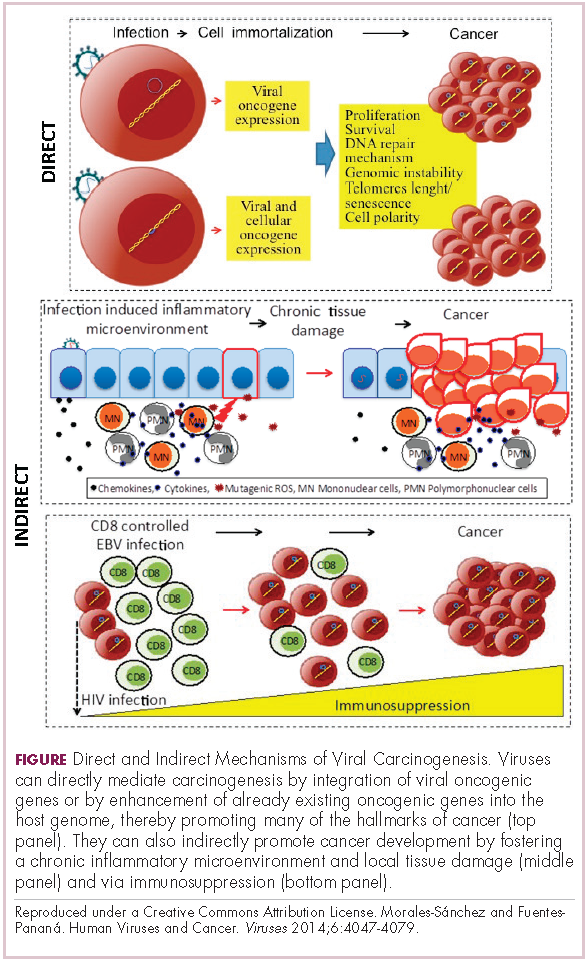
Game changers in pediatric cancer
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
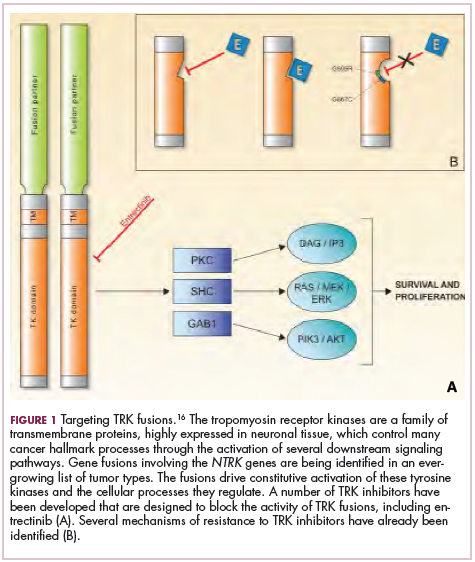
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.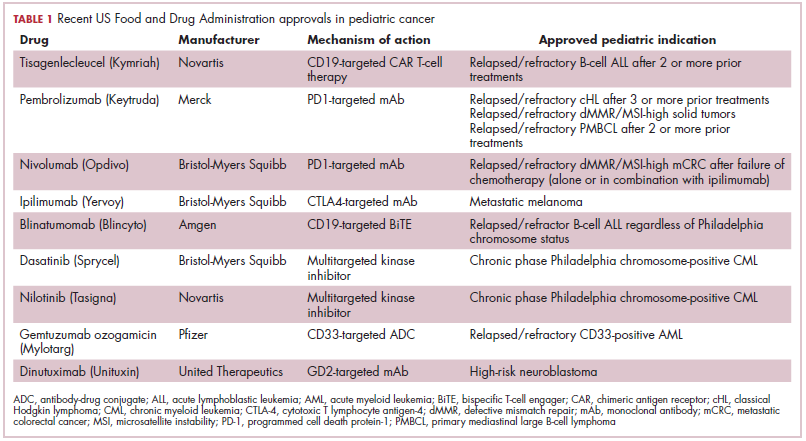
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).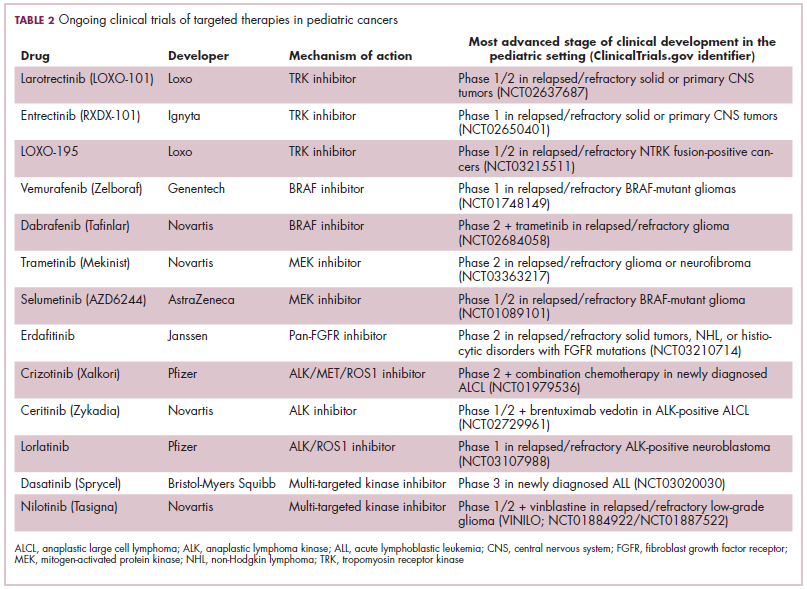
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.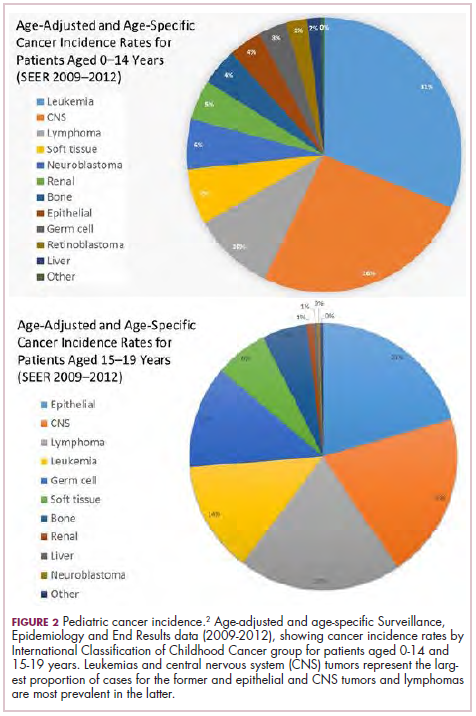
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).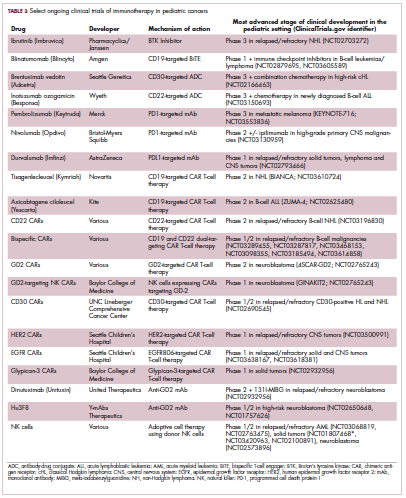
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.