User login
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
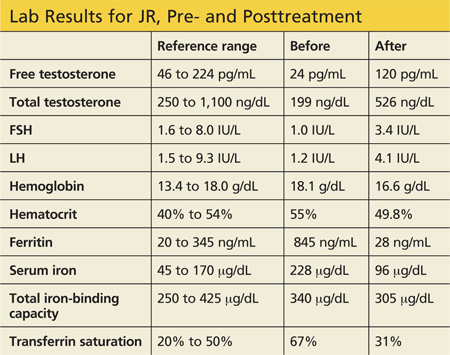
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.