User login
Noninsulinoma Pancreatogenous Hypoglycemia Syndrome Following Gastric Bypass Surgery
A 28-year-old white woman, KR, presents to primary care with episodic diaphoresis and weakness that occur one to two hours after meals. There is no history of syncope or seizures. The hypoglycemic symptoms abate with intake of oral glucose and do not occur when the patient fasts.
KR underwent Roux-en-Y gastric bypass surgery 12 months ago. At the time, her body weight was 250 lbs and her height, 62 in (BMI, 46). She has lost 60 lbs since surgery (current BMI, 35). KR has no comorbid medical conditions. She denies use of insulin injection or oral hypoglycemic medication, as well as alcohol consumption. There is no history of diarrhea or abdominal pain. Her only medication is a daily multivitamin.
Physical exam reveals a blood pressure of 126/80 mm Hg; pulse, 82 beats/min; respiratory rate, 16 breaths/min; and O2 saturation, 98%. Heart rate is regular with no murmur. Lungs are clear to auscultation. Abdominal and neurologic exams are unremarkable; musculoskeletal strength and orthostatic vital signs are normal.
The patient is instructed to test her blood sugar with a glucometer and return to the clinic in two weeks. Fingerstick monitoring reveals that her serum glucose level drops into the 40 to 50 mg/dL range approximately one to two hours after meals containing > 45 g of carbohydrate. Her fasting serum glucose readings are in the 80 to 95 mg/dL range.
The patient is presumptively diagnosed with dumping syndrome and receives nutritional counseling; she is instructed to reduce intake of simple carbohydrates and increase the protein content of meals. Despite these dietary modifications, the episodes of hypoglycemia persist.
The patient is then referred to endocrinology. Fasting labwork reveals a serum glucose level of 85 mg/dL; normal adrenocorticotropic hormone (ACTH) and cortisol levels; C-peptide level, 2.46 ng/mL (reference range, 0.80–4.00 ng/mL); and insulin level, 6.4 mIU/mL (reference range 2.6–24.9 mIU/mL). A 75-g two-hour oral glucose tolerance test (OGTT) reveals peak serum glucose of 180 mg/dL at 30 minutes followed by a nadir serum glucose of 48 mg/dL at 110 minutes, accompanied by hypoglycemic symptoms. The insulin and C-peptide levels are elevated during the entire two-hour test. The serum cortisol level is 22 mg/dL when the glucose level is 48 mg/dL. CT of the abdomen, previously ordered by the patient’s primary care provider, was unremarkable.
Since there is no laboratory evidence of fasting hypoglycemia and no pancreatic abnormalities are seen on imaging studies, the possibility of insulinoma is excluded from the differential diagnosis. Adrenal insufficiency is excluded based on the normal ACTH and cortisol levels. The possibility of noninsulinoma pancreatogenous hypoglycemia syndrome is considered.
The patient is prescribed verapamil ER 100 mg/d and notes significant reduction in the frequency of hypoglycemic episodes and symptoms. She is scheduled for follow-up in four weeks to assess for any changes in the frequency or severity of her hypoglycemic episodes.
BACKGROUND
Postprandial hypoglycemia is a rare but potentially serious complication of bariatric surgery procedures that divert nutrients into the small bowel.1,2 The Bariatric Outcomes Longitudinal Database revealed a 0.1% incidence of hypoglycemia in patients who underwent Roux-en-Y gastric bypass surgery.3
The most common cause of hypoglycemia following gastric bypass surgery is dumping syndrome, which involves rapid emptying of gastric contents with reactive hypoglycemia due to increased postprandial insulin release. In dumping syndrome, hypoglycemic symptoms—flushing, diaphoresis, weakness, and dizziness—typically occur within two to three hours after meals; patients do not experience the more severe symptoms of neuroglycopenia (eg, cognitive impairment, seizures, and loss of consciousness).4 The symptoms of dumping syndrome typically improve with reduced intake of simple carbohydrates and increased protein consumption.1
Other causes of postprandial hypoglycemia include insulinoma and noninsulinoma pancreatogenous hypoglycemia syndrome (NIPHS). Although both diagnoses are rare, they should be considered if no improvement in hypoglycemic symptoms occurs after dietary modification.1
Insulinoma is the most common cause of persistent hyperinsulinemic hypoglycemia. It is defined by Whipple’s triad: symptomatic hypoglycemia during fasting, a serum glucose level > 50 mg/dL at the time of symptom onset, and relief of symptoms after administration of glucose.5
NIPHS is less common than insulinoma. It is characterized by postprandial hypoglycemia due to increased insulin secretion resulting from pancreatic b-cell hyperplasia. Hypoglycemia does not typically occur during a 72-hour fast. In addition, pancreatic imaging studies yield normal results in cases of NIPHS. The selective arterial calcium stimulation test is positive in NIPHS.5 NIPHS is definitively diagnosed by histopathologic examination of the pancreas, which reveals nesidioblastosis.6
Nesidioblastosis involves pathologic b-cell overgrowth in the pancreas that results in excess insulin secretion.4 Nesidioblastosis is characterized by pancreatic b-cell hypertrophy, islet hyperplasia, and increased b-cell mass.2
Nesidioblastosis is the leading cause of hyperinsulinemia in newborns and infants (annual incidence, 1 in 50,000 births) but is quite rare in adults, occurring in 0.5% to 7.0% of all those with hyperinsulinism.7,8 Islet cell hypertrophy—characteristic of nesidioblastosis—is seen in both adults and children, whereas genetic mutations are present only in infants.7
Although rare in adults, nesidioblastosis is more common in the setting of gastric bypass than in the general population.7 As of 2011, there have been 40 cases of nesidioblastosis in adults who received gastric bypass.2 With the rapid increase in the number of these surgeries performed each year, nesidioblastosis should be considered in the differential diagnosis for patients who experience hypoglycemia following the procedure.2,7
Continue for hormonal mechanisms >>
HORMONAL MECHANISMS
There are multiple theories regarding the etiology of b-cell hyperplasia following bariatric surgery. The specific causes for NIPHS after gastric bypass remain under investigation.2
The most common theory is that b-cell hyperplasia may occur as a result of the surgical procedure itself and not due to obesity. The rapid delivery of food to the distal ileum after gastric bypass surgery may result in elevated production of incretin hormones (eg, GLP-1 and GIP), which increase b-cell proliferation, insulin secretion, and insulin sensitivity.7
Roux-en-Y gastric bypass also impairs ghrelin secretion. Ghrelin normally acts to suppress insulin secretion and directly opposes the action of insulin. Reduced levels of ghrelin may increase the likelihood of hypoglycemia. Other hormones that may contribute to the metabolic effects of bariatric surgery include peptide YY, oxyntomodulin, and others as yet unidentified.5,6
CLINICAL MANIFESTATIONS
NIPHS is characterized by moderate to severe postprandial hypoglycemia. Symptoms include confusion, diaphoresis, tremulousness, anxiety, weakness, blurred vision, and disorientation, as well as more severe neuroglycopenic symptoms, such as cognitive impairment, seizures, and loss of consciousness.5
These symptoms do not typically manifest until several months after gastric bypass surgery. (By contrast, symptoms experienced with dumping syndrome typically manifest shortly after the procedure.) Of note, hypoglycemic symptoms of NIPHS do not typically improve after dietary modifications aimed at reducing carbohydrate intake.2
DIAGNOSIS
Diagnosis of NIPHS is based on hypoglycemic/neuroglycopenic signs and symptoms without fasting hypoglycemia; endogenous hyperinsulinemia in the presence of hypoglycemia; negative localization studies for insulinoma (using triple-phase spiral CT); and positive selective arterial calcium stimulation test.4,6
If fasting hypoglycemia is reported or suspected, the patient should be evaluated for insulinoma using a 72-hour fast. During it, glucose, insulin, C-peptide, and pro-insulin levels should be tested every six hours; results will be normal in patients with NIPHS.5
The use of OGTT is controversial, as patients can experience variable degrees of postprandial hyperinsulinism and symptomatic hypoglycemia during the test. There are no guidelines on whether to perform OGTT in the work-up for NIPHS. In research protocols, it is common to perform a five-hour OGTT; subjects consume a mixed meal containing 50 g of carbohydrates, then their glucose, insulin, and C-peptide levels are tested every 30 to 60 minutes (or sooner if hypoglycemic symptoms occur).
Elevated insulin and C-peptide levels in the setting of hypoglycemia are characteristic findings in patients with NIPHS.5,9 In the setting of hypoglycemia, a cortisol level > 20 mg/dL is considered an appropriate adrenal response and excludes adrenal insufficiency. Triple-phase CT of the abdomen should be performed to rule out insulinoma if strongly suspected and if work-up for NIPHS is negative.5
The selective arterial calcium stimulation test is employed to confirm the diagnosis of NIPHS and to guide the extent of pancreatic resection, in an effort to minimize postoperative complications of insulin-dependent diabetes and exocrine insufficiency. In this procedure, the splenic, gastroduodenal, superior mesenteric, and hepatic arteries that supply the pancreas are selectively injected with calcium gluconate. After injection of calcium, the insulin level is measured within each artery.4,5,7 The selective arterial calcium stimulation test can also be used to localize an insulinoma. NIPHS is distinguished from insulinoma by a diffuse increase in insulin secreted from multiple segments of the arteries that supply the pancreas, following calcium stimulation.4,5,7
Continue for treatment >>
TREATMENT
There is no consensus on treatment of NIPHS in postbariatric surgery patients, and no “gold standard” exists. Pharmacologic treatment is recommended prior to surgical intervention in patients who present with symptomatic hypoglycemia without loss of consciousness or seizures.1
Pharmacologic treatments include calcium channel blockers (eg, verapamil or nifedipine), the b-cell inhibitor diazoxide, the secretory inhibitor octreotide, and a-glucosidase inhibitors.1 In one hospital group, patients were initially treated with verapamil ER 100 mg/d.5 If patients did not respond to this therapy or developed adverse effects, diazoxide was added (starting dose, 25 mg tid, titrated to 75 mg tid).5 If this combination did not produce results, octreotide (dose ranging from 25 mg/d to 50 mg tid, subcutaneously) was added. Acarbose can also be added, with the typical starting dose of 50 mg tid.1
Distal or subtotal pancreatectomy to debulk the hypertrophic islets is the most common surgical method used in patients with severe hypoglycemia that is refractory to medical management.2,5 The extent of pancreatic resection is guided by calcium angiography and typically ranges from 80% to 95%.7 Smaller pancreatic resection is associated with higher risk for persistent postoperative hypoglycemia.5 Complications associated with pancreatectomy include insulin-dependent diabetes and exocrine insufficiency.5
It is not uncommon for patients to experience recurrent symptoms after subtotal pancreatectomy, but the symptoms are typically easier to manage pharmacologically than they were pre-operatively. Occasionally, a second surgery with 95% to complete pancreatectomy is employed if recurrent hypoglycemia develops that is refractory to medical management.5
Reversal of Roux-en-Y bypass surgery has been described as an attempted treatment method in several case reports of patients with NIPHS. In at least one patient, hyperinsulinemic hypoglycemia persisted after Roux-en-Y gastric bypass reversal.2 Adjustable gastric band placement was recently reported to reverse hypoglycemic symptoms and maintain weight loss, due to restricted gastric emptying.2 Conversion of Roux-en-Y gastric banding to gastric sleeve may also be employed to restore normal gastrointestinal continuity and resolve hypoglycemia, though limited data is available regarding the efficacy of this procedure.2
Close monitoring is necessary in patients treated with pharmacologic therapy to ensure that symptoms are well controlled and that surgery is not necessary.1
SUMMARY AND CONCLUSION
Symptomatic hypoglycemia is a potential complication associated with gastric bypass surgery and is most commonly caused by dumping syndrome. It is important to consider other causes of postprandial hypoglycemia, such as insulinoma and NIPHS, in patients who continue to experience hypoglycemia despite making dietary modifications.1,4
NIPHS is a rare and poorly understood complication of gastric bypass surgery involving pathologic b-cell overgrowth, leading to hyperinsulinemia and potentially severe hypoglycemia.6 Some patients may present with complete relief of symptoms with pharmacologic treatment, while others will need surgical treatment with subtotal pancreatectomy.1
The findings of increased levels of GLP-1 hormone in patients who have received gastric bypass surgery and the fact that only a very small subset of gastric bypass patients develop NIPHS with histologic features of nesidioblastosis are subjects for further research. Further understanding of the hormonal factors involved in the pathogenesis of NIPHS and adult-onset nesidioblastosis following gastric bypass surgery could lead to novel drug development to treat diabetes.6
REFERENCES
1. Moreira RO, Moreira RBM, Machado NAM, et al. Post-prandial hypoglycemia after bariatric surgery: pharmacological treatment with verapamil and acarbose. Obes Surg. 2008;18:1618-1621.
2. Cui Y, Elahi D, Andersen D. Advances in the etiology and management of hyperinsulinemic hypoglycemia after Roux-en-Y gastric bypass. J Gastrointest Surg. 2011;15:1879-1888.
3. Sarwar H, Chapman III WH, Pender JR, et al. Hypoglycemia after Roux-en-Y gastric bypass: the BOLD experience. Obes Surg. 2014; 24(7):1120-1124.
4. Service GJ, Thompson GB, Service FJ, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353(3):249-254.
5. Mathavan VK, Arregui M, Davis C, et al. Management of postgastric bypass noninsulinoma pancreatogenous hypoglycemia. Surg Endosc. 2010;24:2547-2555.
6. Cummings D. Gastric bypass and nesidioblastosis—too much of a good thing for islets? N Engl J Med. 2005;353(3):300-302.
7. Clancy TE, Moore FD, Zinner MJ. Post-gastric bypass hyperinsulinism with nesidioblastosis: subtotal or total pancreatectomy may be needed to prevent recurrent hypoglycemia. J Gastrointest Surg. 2006;10(8):1116-1119.
8. Kaczirek K, Niederle B. Nesidioblastosis: an old term and a new understanding. World J Surg. 2004;28:1227-1230.
9. Salehi M, Gastaldelli A, D’Alessio DA. Altered islet function and insulin clearance cause hyperinsulinemia in gastric bypass patients with symptoms of postprandial hypoglycemia. J Clin Endocrinol Metab. 2014;99(6): 2008-2017.
A 28-year-old white woman, KR, presents to primary care with episodic diaphoresis and weakness that occur one to two hours after meals. There is no history of syncope or seizures. The hypoglycemic symptoms abate with intake of oral glucose and do not occur when the patient fasts.
KR underwent Roux-en-Y gastric bypass surgery 12 months ago. At the time, her body weight was 250 lbs and her height, 62 in (BMI, 46). She has lost 60 lbs since surgery (current BMI, 35). KR has no comorbid medical conditions. She denies use of insulin injection or oral hypoglycemic medication, as well as alcohol consumption. There is no history of diarrhea or abdominal pain. Her only medication is a daily multivitamin.
Physical exam reveals a blood pressure of 126/80 mm Hg; pulse, 82 beats/min; respiratory rate, 16 breaths/min; and O2 saturation, 98%. Heart rate is regular with no murmur. Lungs are clear to auscultation. Abdominal and neurologic exams are unremarkable; musculoskeletal strength and orthostatic vital signs are normal.
The patient is instructed to test her blood sugar with a glucometer and return to the clinic in two weeks. Fingerstick monitoring reveals that her serum glucose level drops into the 40 to 50 mg/dL range approximately one to two hours after meals containing > 45 g of carbohydrate. Her fasting serum glucose readings are in the 80 to 95 mg/dL range.
The patient is presumptively diagnosed with dumping syndrome and receives nutritional counseling; she is instructed to reduce intake of simple carbohydrates and increase the protein content of meals. Despite these dietary modifications, the episodes of hypoglycemia persist.
The patient is then referred to endocrinology. Fasting labwork reveals a serum glucose level of 85 mg/dL; normal adrenocorticotropic hormone (ACTH) and cortisol levels; C-peptide level, 2.46 ng/mL (reference range, 0.80–4.00 ng/mL); and insulin level, 6.4 mIU/mL (reference range 2.6–24.9 mIU/mL). A 75-g two-hour oral glucose tolerance test (OGTT) reveals peak serum glucose of 180 mg/dL at 30 minutes followed by a nadir serum glucose of 48 mg/dL at 110 minutes, accompanied by hypoglycemic symptoms. The insulin and C-peptide levels are elevated during the entire two-hour test. The serum cortisol level is 22 mg/dL when the glucose level is 48 mg/dL. CT of the abdomen, previously ordered by the patient’s primary care provider, was unremarkable.
Since there is no laboratory evidence of fasting hypoglycemia and no pancreatic abnormalities are seen on imaging studies, the possibility of insulinoma is excluded from the differential diagnosis. Adrenal insufficiency is excluded based on the normal ACTH and cortisol levels. The possibility of noninsulinoma pancreatogenous hypoglycemia syndrome is considered.
The patient is prescribed verapamil ER 100 mg/d and notes significant reduction in the frequency of hypoglycemic episodes and symptoms. She is scheduled for follow-up in four weeks to assess for any changes in the frequency or severity of her hypoglycemic episodes.
BACKGROUND
Postprandial hypoglycemia is a rare but potentially serious complication of bariatric surgery procedures that divert nutrients into the small bowel.1,2 The Bariatric Outcomes Longitudinal Database revealed a 0.1% incidence of hypoglycemia in patients who underwent Roux-en-Y gastric bypass surgery.3
The most common cause of hypoglycemia following gastric bypass surgery is dumping syndrome, which involves rapid emptying of gastric contents with reactive hypoglycemia due to increased postprandial insulin release. In dumping syndrome, hypoglycemic symptoms—flushing, diaphoresis, weakness, and dizziness—typically occur within two to three hours after meals; patients do not experience the more severe symptoms of neuroglycopenia (eg, cognitive impairment, seizures, and loss of consciousness).4 The symptoms of dumping syndrome typically improve with reduced intake of simple carbohydrates and increased protein consumption.1
Other causes of postprandial hypoglycemia include insulinoma and noninsulinoma pancreatogenous hypoglycemia syndrome (NIPHS). Although both diagnoses are rare, they should be considered if no improvement in hypoglycemic symptoms occurs after dietary modification.1
Insulinoma is the most common cause of persistent hyperinsulinemic hypoglycemia. It is defined by Whipple’s triad: symptomatic hypoglycemia during fasting, a serum glucose level > 50 mg/dL at the time of symptom onset, and relief of symptoms after administration of glucose.5
NIPHS is less common than insulinoma. It is characterized by postprandial hypoglycemia due to increased insulin secretion resulting from pancreatic b-cell hyperplasia. Hypoglycemia does not typically occur during a 72-hour fast. In addition, pancreatic imaging studies yield normal results in cases of NIPHS. The selective arterial calcium stimulation test is positive in NIPHS.5 NIPHS is definitively diagnosed by histopathologic examination of the pancreas, which reveals nesidioblastosis.6
Nesidioblastosis involves pathologic b-cell overgrowth in the pancreas that results in excess insulin secretion.4 Nesidioblastosis is characterized by pancreatic b-cell hypertrophy, islet hyperplasia, and increased b-cell mass.2
Nesidioblastosis is the leading cause of hyperinsulinemia in newborns and infants (annual incidence, 1 in 50,000 births) but is quite rare in adults, occurring in 0.5% to 7.0% of all those with hyperinsulinism.7,8 Islet cell hypertrophy—characteristic of nesidioblastosis—is seen in both adults and children, whereas genetic mutations are present only in infants.7
Although rare in adults, nesidioblastosis is more common in the setting of gastric bypass than in the general population.7 As of 2011, there have been 40 cases of nesidioblastosis in adults who received gastric bypass.2 With the rapid increase in the number of these surgeries performed each year, nesidioblastosis should be considered in the differential diagnosis for patients who experience hypoglycemia following the procedure.2,7
Continue for hormonal mechanisms >>
HORMONAL MECHANISMS
There are multiple theories regarding the etiology of b-cell hyperplasia following bariatric surgery. The specific causes for NIPHS after gastric bypass remain under investigation.2
The most common theory is that b-cell hyperplasia may occur as a result of the surgical procedure itself and not due to obesity. The rapid delivery of food to the distal ileum after gastric bypass surgery may result in elevated production of incretin hormones (eg, GLP-1 and GIP), which increase b-cell proliferation, insulin secretion, and insulin sensitivity.7
Roux-en-Y gastric bypass also impairs ghrelin secretion. Ghrelin normally acts to suppress insulin secretion and directly opposes the action of insulin. Reduced levels of ghrelin may increase the likelihood of hypoglycemia. Other hormones that may contribute to the metabolic effects of bariatric surgery include peptide YY, oxyntomodulin, and others as yet unidentified.5,6
CLINICAL MANIFESTATIONS
NIPHS is characterized by moderate to severe postprandial hypoglycemia. Symptoms include confusion, diaphoresis, tremulousness, anxiety, weakness, blurred vision, and disorientation, as well as more severe neuroglycopenic symptoms, such as cognitive impairment, seizures, and loss of consciousness.5
These symptoms do not typically manifest until several months after gastric bypass surgery. (By contrast, symptoms experienced with dumping syndrome typically manifest shortly after the procedure.) Of note, hypoglycemic symptoms of NIPHS do not typically improve after dietary modifications aimed at reducing carbohydrate intake.2
DIAGNOSIS
Diagnosis of NIPHS is based on hypoglycemic/neuroglycopenic signs and symptoms without fasting hypoglycemia; endogenous hyperinsulinemia in the presence of hypoglycemia; negative localization studies for insulinoma (using triple-phase spiral CT); and positive selective arterial calcium stimulation test.4,6
If fasting hypoglycemia is reported or suspected, the patient should be evaluated for insulinoma using a 72-hour fast. During it, glucose, insulin, C-peptide, and pro-insulin levels should be tested every six hours; results will be normal in patients with NIPHS.5
The use of OGTT is controversial, as patients can experience variable degrees of postprandial hyperinsulinism and symptomatic hypoglycemia during the test. There are no guidelines on whether to perform OGTT in the work-up for NIPHS. In research protocols, it is common to perform a five-hour OGTT; subjects consume a mixed meal containing 50 g of carbohydrates, then their glucose, insulin, and C-peptide levels are tested every 30 to 60 minutes (or sooner if hypoglycemic symptoms occur).
Elevated insulin and C-peptide levels in the setting of hypoglycemia are characteristic findings in patients with NIPHS.5,9 In the setting of hypoglycemia, a cortisol level > 20 mg/dL is considered an appropriate adrenal response and excludes adrenal insufficiency. Triple-phase CT of the abdomen should be performed to rule out insulinoma if strongly suspected and if work-up for NIPHS is negative.5
The selective arterial calcium stimulation test is employed to confirm the diagnosis of NIPHS and to guide the extent of pancreatic resection, in an effort to minimize postoperative complications of insulin-dependent diabetes and exocrine insufficiency. In this procedure, the splenic, gastroduodenal, superior mesenteric, and hepatic arteries that supply the pancreas are selectively injected with calcium gluconate. After injection of calcium, the insulin level is measured within each artery.4,5,7 The selective arterial calcium stimulation test can also be used to localize an insulinoma. NIPHS is distinguished from insulinoma by a diffuse increase in insulin secreted from multiple segments of the arteries that supply the pancreas, following calcium stimulation.4,5,7
Continue for treatment >>
TREATMENT
There is no consensus on treatment of NIPHS in postbariatric surgery patients, and no “gold standard” exists. Pharmacologic treatment is recommended prior to surgical intervention in patients who present with symptomatic hypoglycemia without loss of consciousness or seizures.1
Pharmacologic treatments include calcium channel blockers (eg, verapamil or nifedipine), the b-cell inhibitor diazoxide, the secretory inhibitor octreotide, and a-glucosidase inhibitors.1 In one hospital group, patients were initially treated with verapamil ER 100 mg/d.5 If patients did not respond to this therapy or developed adverse effects, diazoxide was added (starting dose, 25 mg tid, titrated to 75 mg tid).5 If this combination did not produce results, octreotide (dose ranging from 25 mg/d to 50 mg tid, subcutaneously) was added. Acarbose can also be added, with the typical starting dose of 50 mg tid.1
Distal or subtotal pancreatectomy to debulk the hypertrophic islets is the most common surgical method used in patients with severe hypoglycemia that is refractory to medical management.2,5 The extent of pancreatic resection is guided by calcium angiography and typically ranges from 80% to 95%.7 Smaller pancreatic resection is associated with higher risk for persistent postoperative hypoglycemia.5 Complications associated with pancreatectomy include insulin-dependent diabetes and exocrine insufficiency.5
It is not uncommon for patients to experience recurrent symptoms after subtotal pancreatectomy, but the symptoms are typically easier to manage pharmacologically than they were pre-operatively. Occasionally, a second surgery with 95% to complete pancreatectomy is employed if recurrent hypoglycemia develops that is refractory to medical management.5
Reversal of Roux-en-Y bypass surgery has been described as an attempted treatment method in several case reports of patients with NIPHS. In at least one patient, hyperinsulinemic hypoglycemia persisted after Roux-en-Y gastric bypass reversal.2 Adjustable gastric band placement was recently reported to reverse hypoglycemic symptoms and maintain weight loss, due to restricted gastric emptying.2 Conversion of Roux-en-Y gastric banding to gastric sleeve may also be employed to restore normal gastrointestinal continuity and resolve hypoglycemia, though limited data is available regarding the efficacy of this procedure.2
Close monitoring is necessary in patients treated with pharmacologic therapy to ensure that symptoms are well controlled and that surgery is not necessary.1
SUMMARY AND CONCLUSION
Symptomatic hypoglycemia is a potential complication associated with gastric bypass surgery and is most commonly caused by dumping syndrome. It is important to consider other causes of postprandial hypoglycemia, such as insulinoma and NIPHS, in patients who continue to experience hypoglycemia despite making dietary modifications.1,4
NIPHS is a rare and poorly understood complication of gastric bypass surgery involving pathologic b-cell overgrowth, leading to hyperinsulinemia and potentially severe hypoglycemia.6 Some patients may present with complete relief of symptoms with pharmacologic treatment, while others will need surgical treatment with subtotal pancreatectomy.1
The findings of increased levels of GLP-1 hormone in patients who have received gastric bypass surgery and the fact that only a very small subset of gastric bypass patients develop NIPHS with histologic features of nesidioblastosis are subjects for further research. Further understanding of the hormonal factors involved in the pathogenesis of NIPHS and adult-onset nesidioblastosis following gastric bypass surgery could lead to novel drug development to treat diabetes.6
REFERENCES
1. Moreira RO, Moreira RBM, Machado NAM, et al. Post-prandial hypoglycemia after bariatric surgery: pharmacological treatment with verapamil and acarbose. Obes Surg. 2008;18:1618-1621.
2. Cui Y, Elahi D, Andersen D. Advances in the etiology and management of hyperinsulinemic hypoglycemia after Roux-en-Y gastric bypass. J Gastrointest Surg. 2011;15:1879-1888.
3. Sarwar H, Chapman III WH, Pender JR, et al. Hypoglycemia after Roux-en-Y gastric bypass: the BOLD experience. Obes Surg. 2014; 24(7):1120-1124.
4. Service GJ, Thompson GB, Service FJ, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353(3):249-254.
5. Mathavan VK, Arregui M, Davis C, et al. Management of postgastric bypass noninsulinoma pancreatogenous hypoglycemia. Surg Endosc. 2010;24:2547-2555.
6. Cummings D. Gastric bypass and nesidioblastosis—too much of a good thing for islets? N Engl J Med. 2005;353(3):300-302.
7. Clancy TE, Moore FD, Zinner MJ. Post-gastric bypass hyperinsulinism with nesidioblastosis: subtotal or total pancreatectomy may be needed to prevent recurrent hypoglycemia. J Gastrointest Surg. 2006;10(8):1116-1119.
8. Kaczirek K, Niederle B. Nesidioblastosis: an old term and a new understanding. World J Surg. 2004;28:1227-1230.
9. Salehi M, Gastaldelli A, D’Alessio DA. Altered islet function and insulin clearance cause hyperinsulinemia in gastric bypass patients with symptoms of postprandial hypoglycemia. J Clin Endocrinol Metab. 2014;99(6): 2008-2017.
A 28-year-old white woman, KR, presents to primary care with episodic diaphoresis and weakness that occur one to two hours after meals. There is no history of syncope or seizures. The hypoglycemic symptoms abate with intake of oral glucose and do not occur when the patient fasts.
KR underwent Roux-en-Y gastric bypass surgery 12 months ago. At the time, her body weight was 250 lbs and her height, 62 in (BMI, 46). She has lost 60 lbs since surgery (current BMI, 35). KR has no comorbid medical conditions. She denies use of insulin injection or oral hypoglycemic medication, as well as alcohol consumption. There is no history of diarrhea or abdominal pain. Her only medication is a daily multivitamin.
Physical exam reveals a blood pressure of 126/80 mm Hg; pulse, 82 beats/min; respiratory rate, 16 breaths/min; and O2 saturation, 98%. Heart rate is regular with no murmur. Lungs are clear to auscultation. Abdominal and neurologic exams are unremarkable; musculoskeletal strength and orthostatic vital signs are normal.
The patient is instructed to test her blood sugar with a glucometer and return to the clinic in two weeks. Fingerstick monitoring reveals that her serum glucose level drops into the 40 to 50 mg/dL range approximately one to two hours after meals containing > 45 g of carbohydrate. Her fasting serum glucose readings are in the 80 to 95 mg/dL range.
The patient is presumptively diagnosed with dumping syndrome and receives nutritional counseling; she is instructed to reduce intake of simple carbohydrates and increase the protein content of meals. Despite these dietary modifications, the episodes of hypoglycemia persist.
The patient is then referred to endocrinology. Fasting labwork reveals a serum glucose level of 85 mg/dL; normal adrenocorticotropic hormone (ACTH) and cortisol levels; C-peptide level, 2.46 ng/mL (reference range, 0.80–4.00 ng/mL); and insulin level, 6.4 mIU/mL (reference range 2.6–24.9 mIU/mL). A 75-g two-hour oral glucose tolerance test (OGTT) reveals peak serum glucose of 180 mg/dL at 30 minutes followed by a nadir serum glucose of 48 mg/dL at 110 minutes, accompanied by hypoglycemic symptoms. The insulin and C-peptide levels are elevated during the entire two-hour test. The serum cortisol level is 22 mg/dL when the glucose level is 48 mg/dL. CT of the abdomen, previously ordered by the patient’s primary care provider, was unremarkable.
Since there is no laboratory evidence of fasting hypoglycemia and no pancreatic abnormalities are seen on imaging studies, the possibility of insulinoma is excluded from the differential diagnosis. Adrenal insufficiency is excluded based on the normal ACTH and cortisol levels. The possibility of noninsulinoma pancreatogenous hypoglycemia syndrome is considered.
The patient is prescribed verapamil ER 100 mg/d and notes significant reduction in the frequency of hypoglycemic episodes and symptoms. She is scheduled for follow-up in four weeks to assess for any changes in the frequency or severity of her hypoglycemic episodes.
BACKGROUND
Postprandial hypoglycemia is a rare but potentially serious complication of bariatric surgery procedures that divert nutrients into the small bowel.1,2 The Bariatric Outcomes Longitudinal Database revealed a 0.1% incidence of hypoglycemia in patients who underwent Roux-en-Y gastric bypass surgery.3
The most common cause of hypoglycemia following gastric bypass surgery is dumping syndrome, which involves rapid emptying of gastric contents with reactive hypoglycemia due to increased postprandial insulin release. In dumping syndrome, hypoglycemic symptoms—flushing, diaphoresis, weakness, and dizziness—typically occur within two to three hours after meals; patients do not experience the more severe symptoms of neuroglycopenia (eg, cognitive impairment, seizures, and loss of consciousness).4 The symptoms of dumping syndrome typically improve with reduced intake of simple carbohydrates and increased protein consumption.1
Other causes of postprandial hypoglycemia include insulinoma and noninsulinoma pancreatogenous hypoglycemia syndrome (NIPHS). Although both diagnoses are rare, they should be considered if no improvement in hypoglycemic symptoms occurs after dietary modification.1
Insulinoma is the most common cause of persistent hyperinsulinemic hypoglycemia. It is defined by Whipple’s triad: symptomatic hypoglycemia during fasting, a serum glucose level > 50 mg/dL at the time of symptom onset, and relief of symptoms after administration of glucose.5
NIPHS is less common than insulinoma. It is characterized by postprandial hypoglycemia due to increased insulin secretion resulting from pancreatic b-cell hyperplasia. Hypoglycemia does not typically occur during a 72-hour fast. In addition, pancreatic imaging studies yield normal results in cases of NIPHS. The selective arterial calcium stimulation test is positive in NIPHS.5 NIPHS is definitively diagnosed by histopathologic examination of the pancreas, which reveals nesidioblastosis.6
Nesidioblastosis involves pathologic b-cell overgrowth in the pancreas that results in excess insulin secretion.4 Nesidioblastosis is characterized by pancreatic b-cell hypertrophy, islet hyperplasia, and increased b-cell mass.2
Nesidioblastosis is the leading cause of hyperinsulinemia in newborns and infants (annual incidence, 1 in 50,000 births) but is quite rare in adults, occurring in 0.5% to 7.0% of all those with hyperinsulinism.7,8 Islet cell hypertrophy—characteristic of nesidioblastosis—is seen in both adults and children, whereas genetic mutations are present only in infants.7
Although rare in adults, nesidioblastosis is more common in the setting of gastric bypass than in the general population.7 As of 2011, there have been 40 cases of nesidioblastosis in adults who received gastric bypass.2 With the rapid increase in the number of these surgeries performed each year, nesidioblastosis should be considered in the differential diagnosis for patients who experience hypoglycemia following the procedure.2,7
Continue for hormonal mechanisms >>
HORMONAL MECHANISMS
There are multiple theories regarding the etiology of b-cell hyperplasia following bariatric surgery. The specific causes for NIPHS after gastric bypass remain under investigation.2
The most common theory is that b-cell hyperplasia may occur as a result of the surgical procedure itself and not due to obesity. The rapid delivery of food to the distal ileum after gastric bypass surgery may result in elevated production of incretin hormones (eg, GLP-1 and GIP), which increase b-cell proliferation, insulin secretion, and insulin sensitivity.7
Roux-en-Y gastric bypass also impairs ghrelin secretion. Ghrelin normally acts to suppress insulin secretion and directly opposes the action of insulin. Reduced levels of ghrelin may increase the likelihood of hypoglycemia. Other hormones that may contribute to the metabolic effects of bariatric surgery include peptide YY, oxyntomodulin, and others as yet unidentified.5,6
CLINICAL MANIFESTATIONS
NIPHS is characterized by moderate to severe postprandial hypoglycemia. Symptoms include confusion, diaphoresis, tremulousness, anxiety, weakness, blurred vision, and disorientation, as well as more severe neuroglycopenic symptoms, such as cognitive impairment, seizures, and loss of consciousness.5
These symptoms do not typically manifest until several months after gastric bypass surgery. (By contrast, symptoms experienced with dumping syndrome typically manifest shortly after the procedure.) Of note, hypoglycemic symptoms of NIPHS do not typically improve after dietary modifications aimed at reducing carbohydrate intake.2
DIAGNOSIS
Diagnosis of NIPHS is based on hypoglycemic/neuroglycopenic signs and symptoms without fasting hypoglycemia; endogenous hyperinsulinemia in the presence of hypoglycemia; negative localization studies for insulinoma (using triple-phase spiral CT); and positive selective arterial calcium stimulation test.4,6
If fasting hypoglycemia is reported or suspected, the patient should be evaluated for insulinoma using a 72-hour fast. During it, glucose, insulin, C-peptide, and pro-insulin levels should be tested every six hours; results will be normal in patients with NIPHS.5
The use of OGTT is controversial, as patients can experience variable degrees of postprandial hyperinsulinism and symptomatic hypoglycemia during the test. There are no guidelines on whether to perform OGTT in the work-up for NIPHS. In research protocols, it is common to perform a five-hour OGTT; subjects consume a mixed meal containing 50 g of carbohydrates, then their glucose, insulin, and C-peptide levels are tested every 30 to 60 minutes (or sooner if hypoglycemic symptoms occur).
Elevated insulin and C-peptide levels in the setting of hypoglycemia are characteristic findings in patients with NIPHS.5,9 In the setting of hypoglycemia, a cortisol level > 20 mg/dL is considered an appropriate adrenal response and excludes adrenal insufficiency. Triple-phase CT of the abdomen should be performed to rule out insulinoma if strongly suspected and if work-up for NIPHS is negative.5
The selective arterial calcium stimulation test is employed to confirm the diagnosis of NIPHS and to guide the extent of pancreatic resection, in an effort to minimize postoperative complications of insulin-dependent diabetes and exocrine insufficiency. In this procedure, the splenic, gastroduodenal, superior mesenteric, and hepatic arteries that supply the pancreas are selectively injected with calcium gluconate. After injection of calcium, the insulin level is measured within each artery.4,5,7 The selective arterial calcium stimulation test can also be used to localize an insulinoma. NIPHS is distinguished from insulinoma by a diffuse increase in insulin secreted from multiple segments of the arteries that supply the pancreas, following calcium stimulation.4,5,7
Continue for treatment >>
TREATMENT
There is no consensus on treatment of NIPHS in postbariatric surgery patients, and no “gold standard” exists. Pharmacologic treatment is recommended prior to surgical intervention in patients who present with symptomatic hypoglycemia without loss of consciousness or seizures.1
Pharmacologic treatments include calcium channel blockers (eg, verapamil or nifedipine), the b-cell inhibitor diazoxide, the secretory inhibitor octreotide, and a-glucosidase inhibitors.1 In one hospital group, patients were initially treated with verapamil ER 100 mg/d.5 If patients did not respond to this therapy or developed adverse effects, diazoxide was added (starting dose, 25 mg tid, titrated to 75 mg tid).5 If this combination did not produce results, octreotide (dose ranging from 25 mg/d to 50 mg tid, subcutaneously) was added. Acarbose can also be added, with the typical starting dose of 50 mg tid.1
Distal or subtotal pancreatectomy to debulk the hypertrophic islets is the most common surgical method used in patients with severe hypoglycemia that is refractory to medical management.2,5 The extent of pancreatic resection is guided by calcium angiography and typically ranges from 80% to 95%.7 Smaller pancreatic resection is associated with higher risk for persistent postoperative hypoglycemia.5 Complications associated with pancreatectomy include insulin-dependent diabetes and exocrine insufficiency.5
It is not uncommon for patients to experience recurrent symptoms after subtotal pancreatectomy, but the symptoms are typically easier to manage pharmacologically than they were pre-operatively. Occasionally, a second surgery with 95% to complete pancreatectomy is employed if recurrent hypoglycemia develops that is refractory to medical management.5
Reversal of Roux-en-Y bypass surgery has been described as an attempted treatment method in several case reports of patients with NIPHS. In at least one patient, hyperinsulinemic hypoglycemia persisted after Roux-en-Y gastric bypass reversal.2 Adjustable gastric band placement was recently reported to reverse hypoglycemic symptoms and maintain weight loss, due to restricted gastric emptying.2 Conversion of Roux-en-Y gastric banding to gastric sleeve may also be employed to restore normal gastrointestinal continuity and resolve hypoglycemia, though limited data is available regarding the efficacy of this procedure.2
Close monitoring is necessary in patients treated with pharmacologic therapy to ensure that symptoms are well controlled and that surgery is not necessary.1
SUMMARY AND CONCLUSION
Symptomatic hypoglycemia is a potential complication associated with gastric bypass surgery and is most commonly caused by dumping syndrome. It is important to consider other causes of postprandial hypoglycemia, such as insulinoma and NIPHS, in patients who continue to experience hypoglycemia despite making dietary modifications.1,4
NIPHS is a rare and poorly understood complication of gastric bypass surgery involving pathologic b-cell overgrowth, leading to hyperinsulinemia and potentially severe hypoglycemia.6 Some patients may present with complete relief of symptoms with pharmacologic treatment, while others will need surgical treatment with subtotal pancreatectomy.1
The findings of increased levels of GLP-1 hormone in patients who have received gastric bypass surgery and the fact that only a very small subset of gastric bypass patients develop NIPHS with histologic features of nesidioblastosis are subjects for further research. Further understanding of the hormonal factors involved in the pathogenesis of NIPHS and adult-onset nesidioblastosis following gastric bypass surgery could lead to novel drug development to treat diabetes.6
REFERENCES
1. Moreira RO, Moreira RBM, Machado NAM, et al. Post-prandial hypoglycemia after bariatric surgery: pharmacological treatment with verapamil and acarbose. Obes Surg. 2008;18:1618-1621.
2. Cui Y, Elahi D, Andersen D. Advances in the etiology and management of hyperinsulinemic hypoglycemia after Roux-en-Y gastric bypass. J Gastrointest Surg. 2011;15:1879-1888.
3. Sarwar H, Chapman III WH, Pender JR, et al. Hypoglycemia after Roux-en-Y gastric bypass: the BOLD experience. Obes Surg. 2014; 24(7):1120-1124.
4. Service GJ, Thompson GB, Service FJ, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353(3):249-254.
5. Mathavan VK, Arregui M, Davis C, et al. Management of postgastric bypass noninsulinoma pancreatogenous hypoglycemia. Surg Endosc. 2010;24:2547-2555.
6. Cummings D. Gastric bypass and nesidioblastosis—too much of a good thing for islets? N Engl J Med. 2005;353(3):300-302.
7. Clancy TE, Moore FD, Zinner MJ. Post-gastric bypass hyperinsulinism with nesidioblastosis: subtotal or total pancreatectomy may be needed to prevent recurrent hypoglycemia. J Gastrointest Surg. 2006;10(8):1116-1119.
8. Kaczirek K, Niederle B. Nesidioblastosis: an old term and a new understanding. World J Surg. 2004;28:1227-1230.
9. Salehi M, Gastaldelli A, D’Alessio DA. Altered islet function and insulin clearance cause hyperinsulinemia in gastric bypass patients with symptoms of postprandial hypoglycemia. J Clin Endocrinol Metab. 2014;99(6): 2008-2017.
Diabetic Amyotrophy: A Rare but Striking Neuropathy
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
Hereditary Hemochromatosis as a Cause of Hypogonadism
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
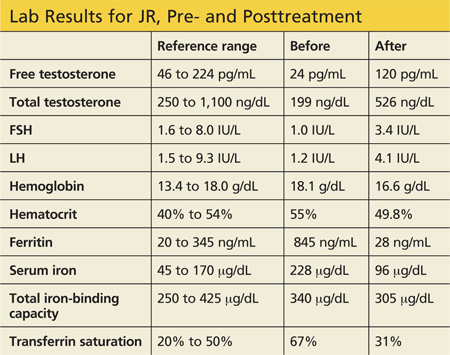
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.