User login
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
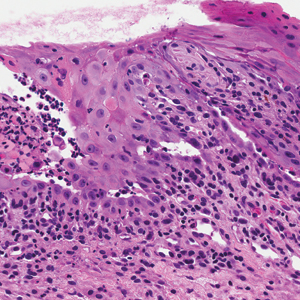
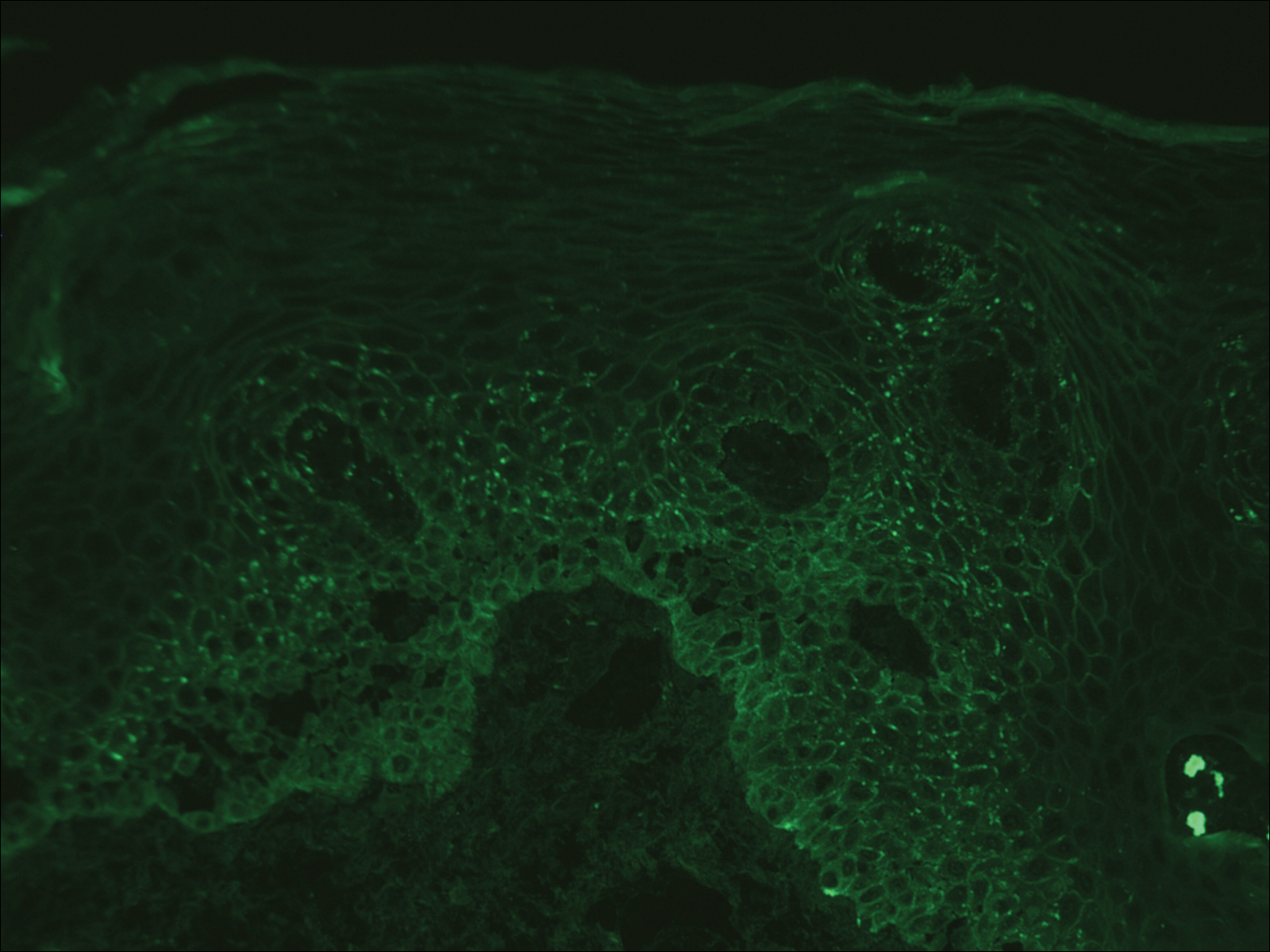
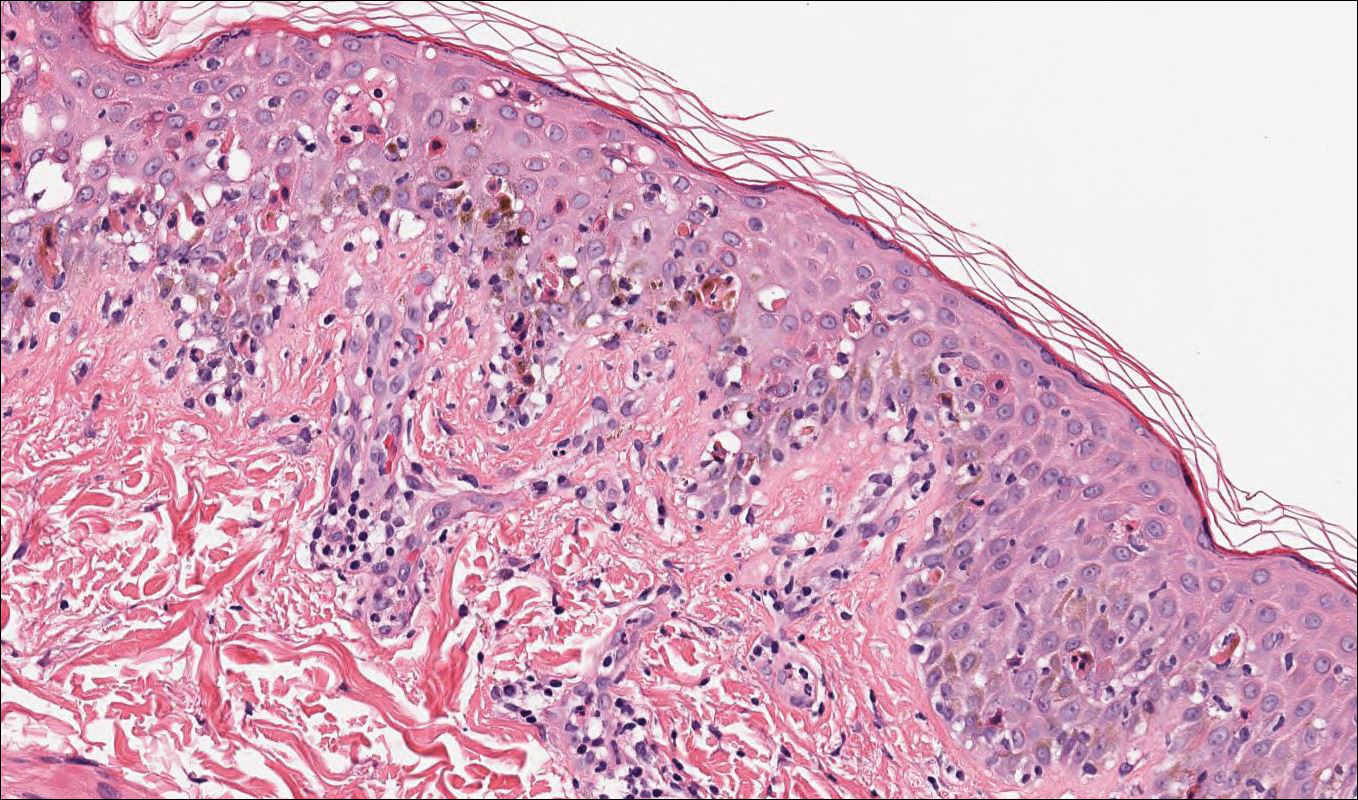
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6

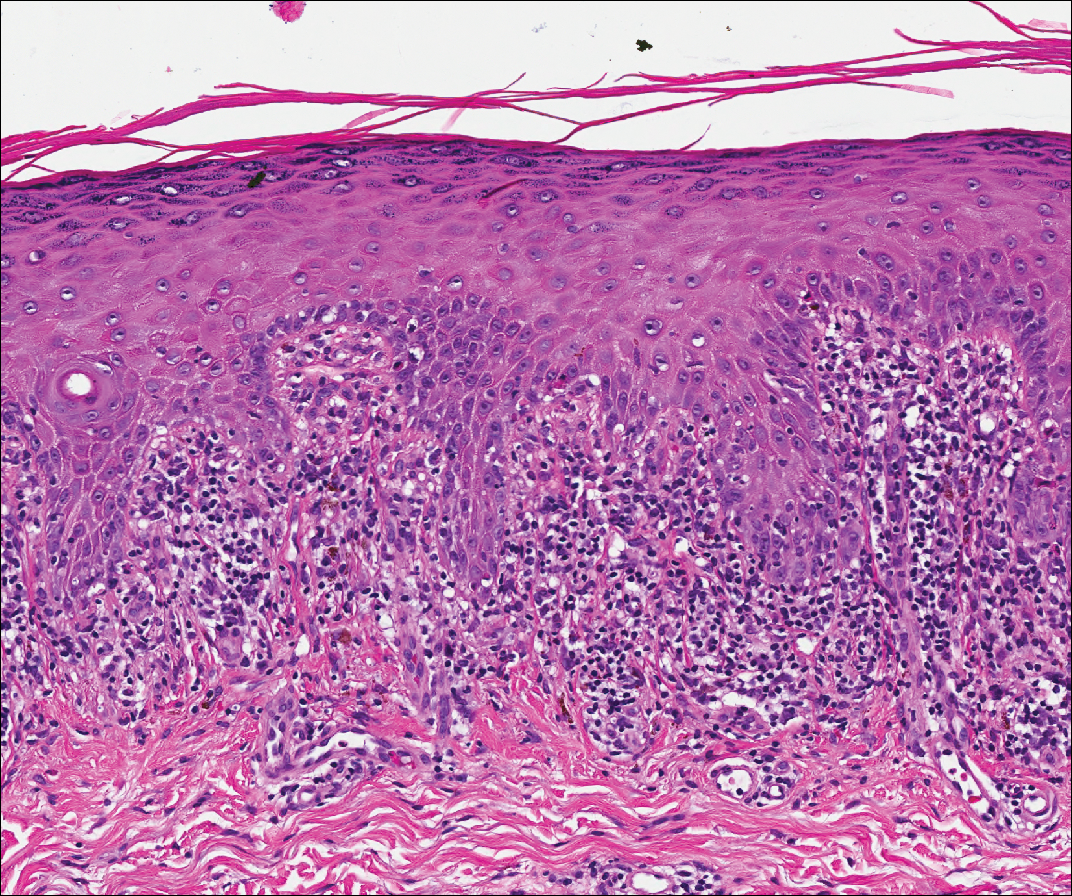
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
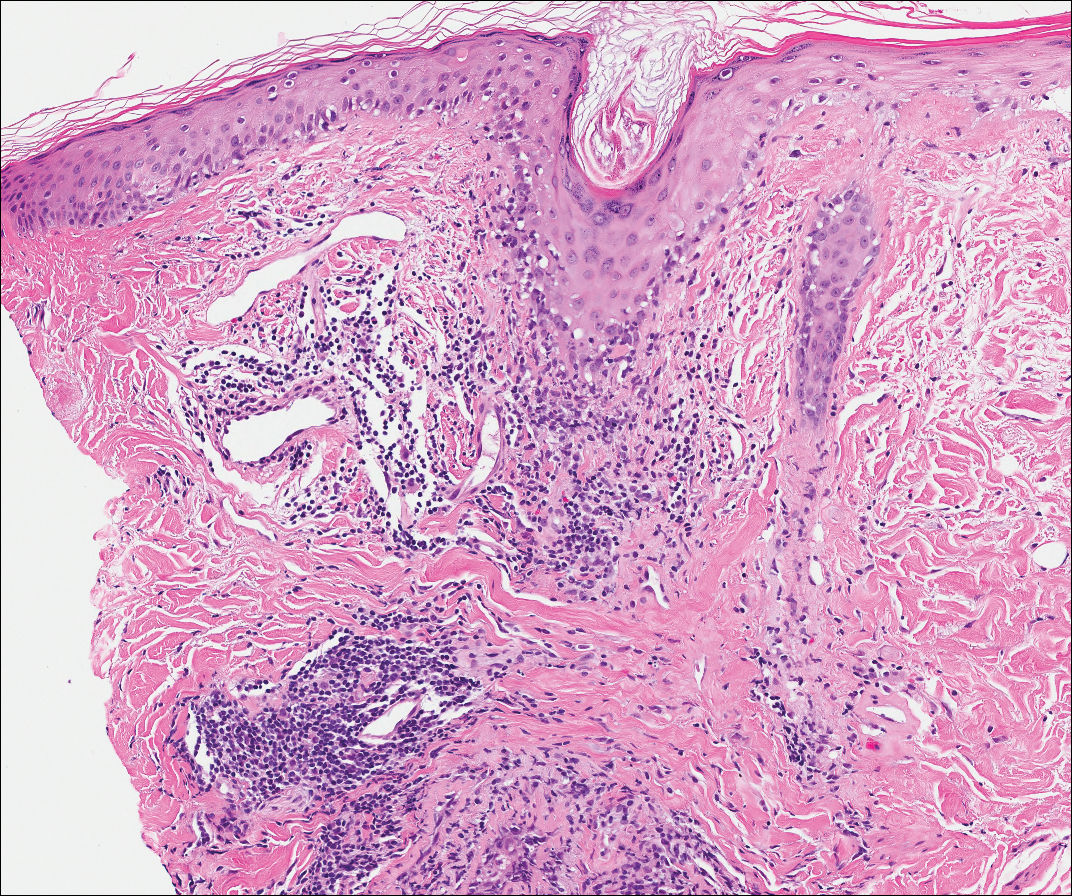
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
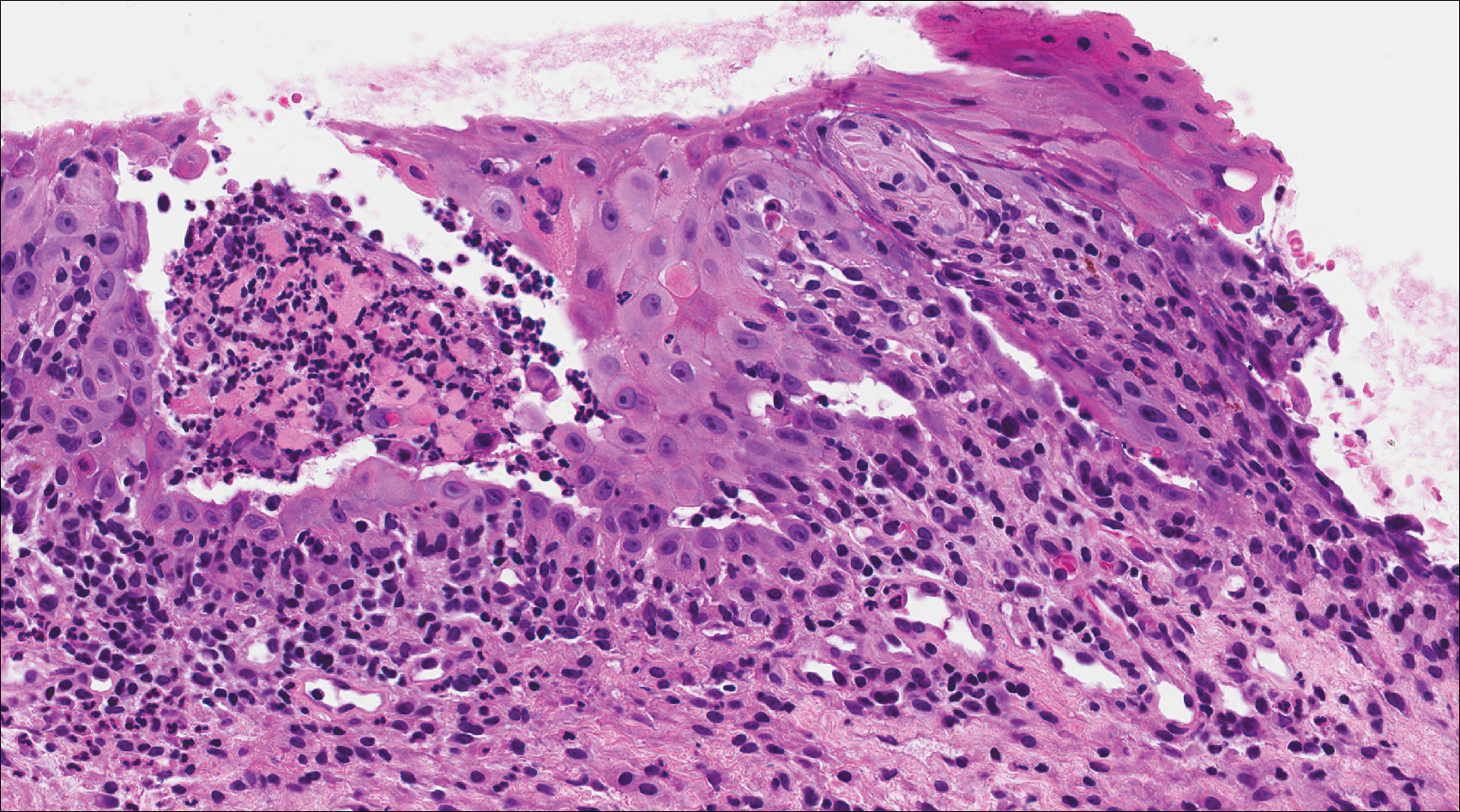
A 41-year-old woman presented with painful ulcers on the oral mucosa of 2 months' duration that were unresponsive to treatment with acyclovir. She had been diagnosed with a pelvic tumor a few weeks prior to the development of the mouth ulcers. Direct immunofluorescence of the perilesional mucosa showed positive IgG and complement C3 with an intercellular distribution. A biopsy of an oral lesion was performed.