User login
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
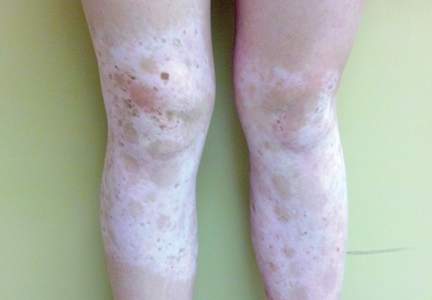
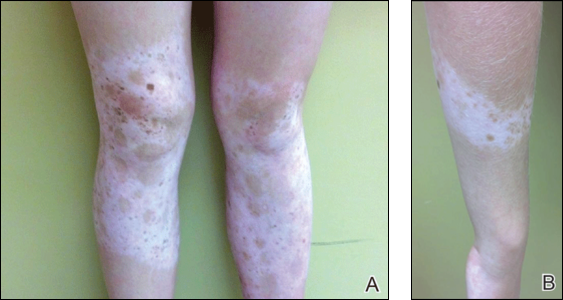
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Practice Points
- Poliosis circumscripta (or white forelock) is commonly the only manifestation of piebaldism in children.
- Affected areas of leukoderma in piebaldism are classically distributed on the central forehead, anterior trunk, and mid extremities.
- The presence of congenital leukoderma should prompt a thorough skin examination and review of the patient’s medical history for evidence of ocular, auditory, and/or neurologic abnormalities.