User login
The Diagnosis: Incontinentia Pigmenti
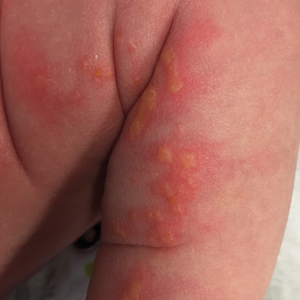
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.

A 4-day-old female neonate presented to the dermatology clinic with a vesicular eruption on the left leg of 1 day's duration. The eruption was asymptomatic without any extracutaneous findings. This term infant was born without complication, and the mother denied any symptoms consistent with herpes simplex virus infection. Physical examination revealed yellow-red vesicles on an erythematous base in a blaschkoid distribution on the left leg. The rest of the examination was unremarkable. Herpes simplex virus polymerase chain reaction testing was negative.