User login
Case
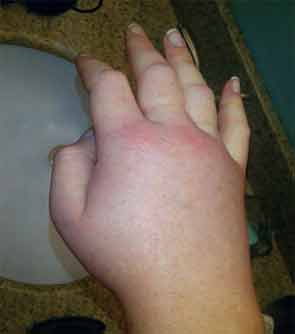
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
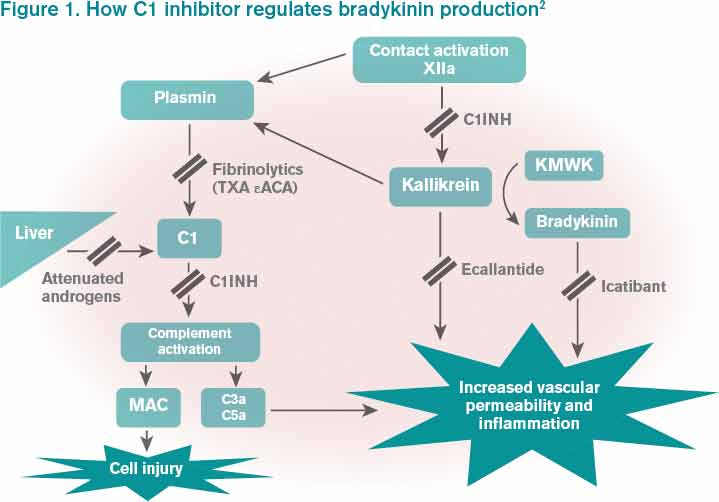
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
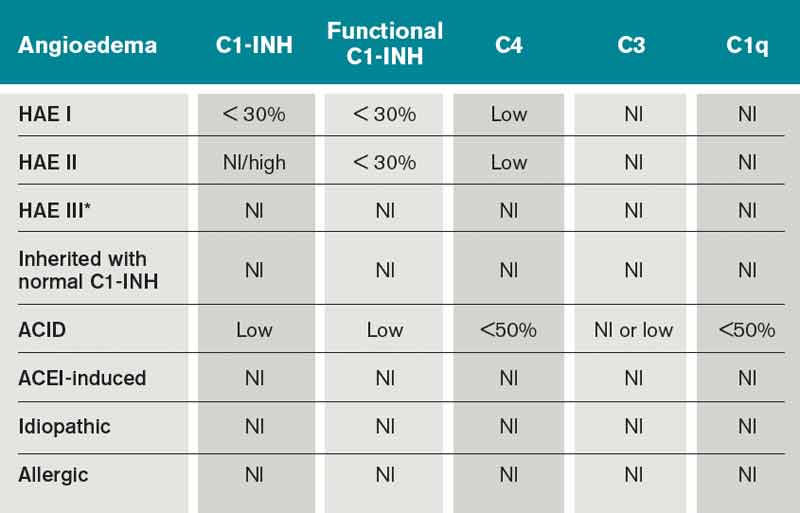
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
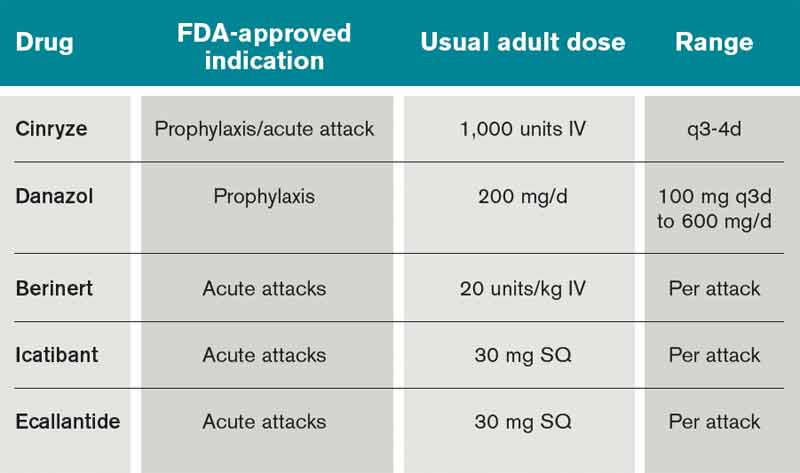
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.
Case
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.
Case
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.