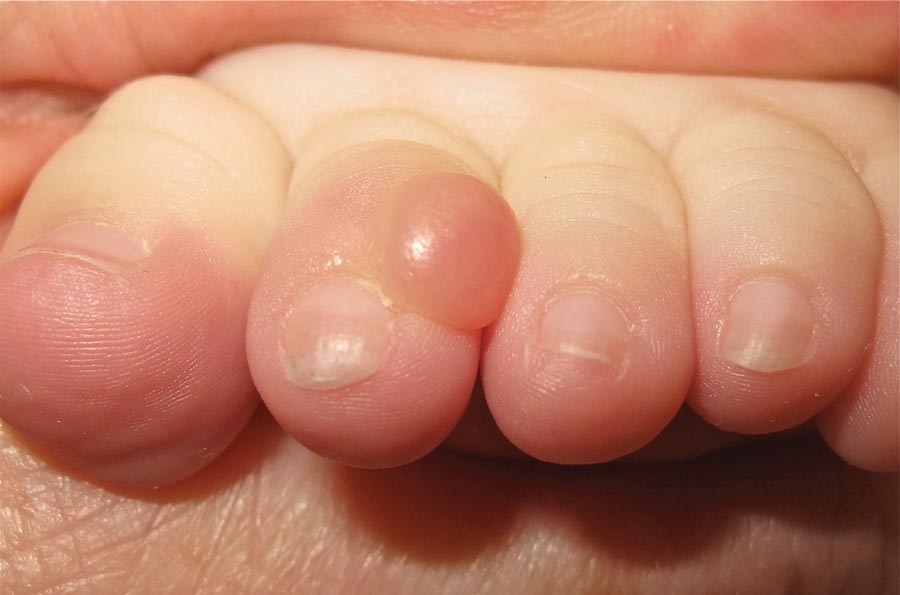
The Diagnosis: Infantile Digital Fibromatosis
On examination, the patient appeared well developed, well nourished, and had a 1×0.5-cm, flesh-colored, firm, nontender nodule on the dorsolateral aspect of the left second toe. After excision by a pediatric surgeon, the specimen was submitted for histopathologic examination. Dense bands of collagen with spindled myofibroblasts containing characteristic eosinophilic cytoplasmic inclusion bodies staining with phosphotungstic acid hematoxylin confirmed the diagnosis of infantile digital fibromatosis (Figure). Postoperatively the patient did well with normal healing and no complications. After 4 months, a recurrence was noted and the parents were considering reexcision.
|
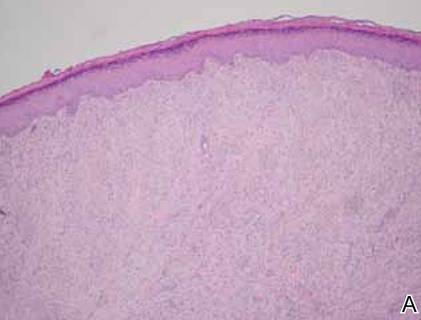
Infantile digital fibromatosis is a rare, benign, often spontaneously regressing, fibrous tissue tumor of infancy and childhood.1 The prevalence of this tumor is unknown. It can be present at birth or more commonly appears in the first year of life. The lesions present as 1- to 2-cm, firm, flesh-colored nodules that initially grow slowly but have the potential for rapid growth in subsequent months. They occur preferentially on the extensor aspects of the digits, typically sparing the thumb and great toe.1 The clinical differential diagnosis includes keloids or hypertrophic scars, granuloma annulare, sarcoidosis, acral fibrokeratomas, periungual fibromas, supernumerary digits, pachydermodactyly, juvenile aponeurotic fibroma, and terminal osseous dysplasia and pigmentary defects.2
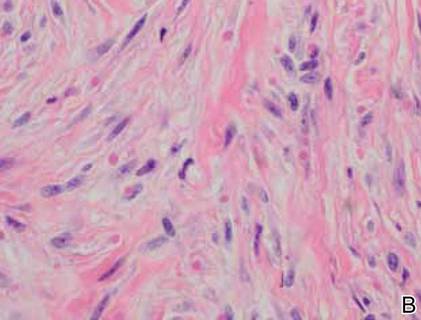
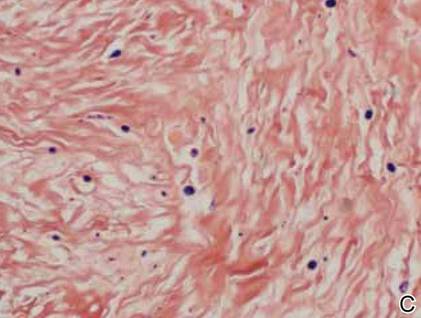
The histology of infantile digital fibromatosis is distinctive. Spindled myofibroblasts that contain round or ovoid, eosinophilic, cytoplasmic inclusion bodies composed of an accumulation of actin and vimentin filaments are characteristic.1 Inclusions are typically juxtanuclear and may indent the adjacent nucleus. The inclusion bodies stain red with Masson trichrome stain and purple with phosphotungstic acid hematoxylin stain.1 The histopathologic differential diagnosis includes scar, angiofibroma, dermatofibroma, neurofibroma, and angiofibromatous verruca vulgaris.
Although many treatments exist for infantile digital fibromatosis, optimal therapy is not standardized. Most lesions spontaneously regress, but func-tional disability with deforming contractures can occur if untreated. Topical therapy with imiquimod cream 5% and diflucortolone valerate cream have been reported to produce no effect on tumor size.3 Intralesional 5-fluorouracil was successful in treating a patient after 5 monthly injections.4 Intralesional triamcinolone 10 mg/cc injections were shown to be a well-tolerated and successful treatment in a case series of 7 patients.5 The most utilized intervention appears to be standard surgery, with a few patients treated with Mohs micrographic surgery.6,7
Treatment with surgical excision often results in recurrence, with studies showing a 50% to 75% recurrence rate.8,9 Our case is not atypical and illustrates this phenomenon. Other reported complications of surgical management include hypertrophic scarring and reduced distal interphalangeal joint mobility.5 Unless infantile digital fibromatosis causes mobility dysfunction or related disabilities, observation with regular follow-up should be considered, as lesions can spontaneously regress.