Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations.
By considering gene interactions, this approach provides the physician with a more nuanced understanding of the prognosis and informs the selection of therapies either at the time of diagnosis or at the time of relapse.9,21,22 Mutations of TP53 and ASXL1, for example, are consistently associated with worse prognosis and are now included along with FLT3, NPM1, CEBPA, and TP53 in the National Comprehensive Cancer Network (NCCN) and European LeukemiaNet (ELN) risk stratification schemas (Table 1).3
Diagnosis and Classification
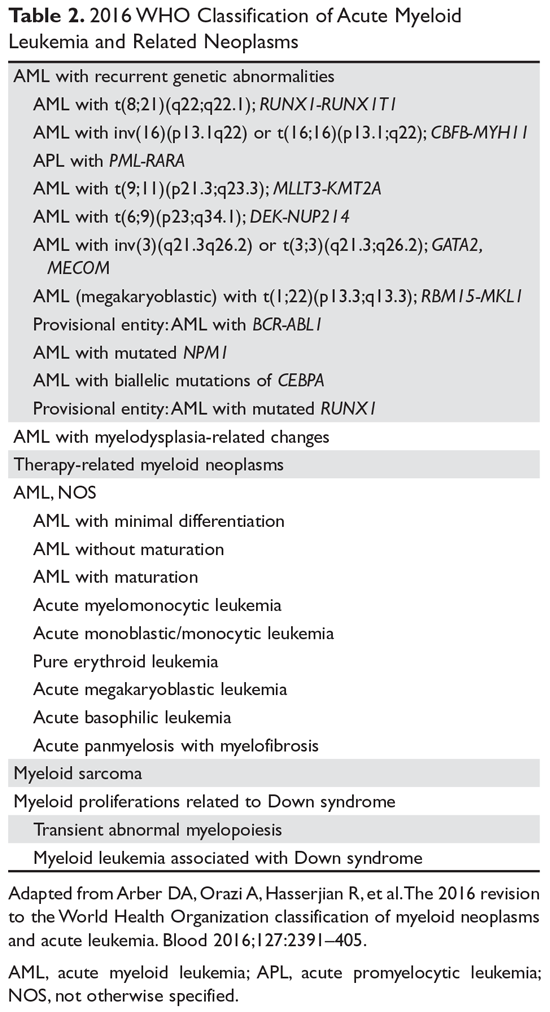
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia.
Six groups of AML are recognized under this scheme. “AML with recurrent genetic abnormalities” accounts for about 20% to 30% of all AML cases and contains the most distinct genetic abnormalities of prognostic significance.19,23 AML with t(8;21) and AML with inv(16) or t(16;16), the 2 forms of core-binding factor AML seen in about 10% to 15% of patients, fall under this group and have a relatively good prognosis. The presence of c-KIT mutation is, however, an adverse prognostic feature in these core-binding factor AMLs.24 Overall, this group includes 8 cytogenetically defined abnormalities, 1 molecular abnormality (AML with mutated NPM1), and 2 other provisional entities (AML with biallelic mutations of CEBPA and AML with mutated RUNX1).