User login
American College of Rheumatology (ACR): Annual Scientific Meeting
Depression accounts for psoriatics’ increased MI risk
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
AT THE ACR ANNUAL MEETING
Major finding: Patients with psoriasis or psoriatic arthritis who developed comorbid depression had a highly significant 80% increased risk of acute MI during follow-up, compared with those without depression.
Data source: This was a population-based cohort study involving 10,041 adults diagnosed with psoriasis or psoriatic arthritis in British Columbia during 1996-2006 and more than 47,000 controls.
Disclosures: The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
Apremilast’s positive study results made it the talk of ACR 2013
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
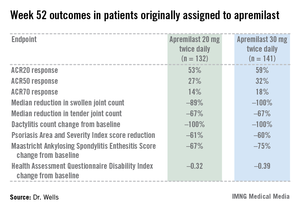
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
AT THE ACR ANNUAL MEETING
Major finding: After 16 weeks of treatment, the investigational drug apremilast achieved an ACR20 response of 29.2% at 20 mg twice daily as first-line therapy in patients with psoriatic arthritis and 32.3% at 30 mg twice daily, whereas placebo-treated controls achieved a rate of 16.9%.
Data source: The PALACE 4 study was a pivotal phase III, randomized, placebo-controlled trial including 527 patients with active psoriatic arthritis.
Disclosures: PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
RA triple therapy wins cost-effectiveness comparison vs. etanercept plus methotrexate
SAN DIEGO – In patients with early rheumatoid arthritis, treatment with a combination of disease-modifying antirheumatic drugs is more cost effective than an immediate or step-up approach using etanercept and methotrexate, a new analysis demonstrated.
"The benefits from all strategies were comparable, but biologics strategies were almost twice as expensive [as] triple strategies, producing incremental cost-effectiveness ratios higher than what most health care settings would find acceptable," Kaleb Michaud, Ph.D., said at the annual meeting of the American College of Rheumatology.

Dr. Michaud of the division of rheumatology and immunology at the University of Nebraska Medical Center, Omaha, and his associates analyzed data from 755 patients enrolled in the randomized, 2-year TEAR (Treatment of Early Aggressive RA) trial (Arthritis Rheum. 2012;64:2824-35), which compared the long-term effectiveness of using a triple therapy approach for treating RA with a strategy of using a etanercept (Enbrel) plus methotrexate. They studied four approaches: immediate triple therapy of methotrexate, sulfasalazine, and hydroxychloroquine; immediate treatment with etanercept and methotrexate; a step-up triple therapy; and a step-up etanercept therapy. The researchers incorporated estimates of annual discontinuation rates of triple therapy and etanercept of 22% and 10%, respectively, based on data from the National Data Bank for Rheumatic Diseases.
The analysis was limited to patients with RA duration of less than 3 years and those with active disease, defined as having four or more swollen joints with seropositivity and/or erosions. The mean disease duration was 3.6 months and the mean Disease Activity Score was 5.8. The researchers used a societal perspective and a Markov cohort model with 3% discounting to estimate quality-adjusted life year (QALY) measurements and the costs associated with therapy approaches in the TEAR trial.
Dr. Michaud, who is also codirector of the National Data Bank for Rheumatic Diseases, reported that the lifetime benefit of all four treatment approaches were comparable, within 0.06 QALYs of each other. However, the therapies that included etanercept were nearly twice as expensive because of the higher cost of the TNF inhibitor. Specifically, the costs were $152,400 in the immediate triple therapy group and $154,900 in the step-up triple therapy group, compared with $269,500 in the step-up etanercept group and $338,100 in the immediate etanercept group. The researchers determined that the lifetime incremental cost-effectiveness ratio of immediate etanercept to immediate triple therapy was $837,100/QALY.
"The immediate triple therapy dominated other arms at 1 and 2 years in this trial, and its results were most sensitive to the costs of etanercept and the reduction in triple therapy discontinuation rates," Dr. Michaud commented. "Our sensitivity analysis did not change our conclusions."
Dr. Michaud disclosed that the study was supported in part by a 2012 investigator award from the Rheumatology Research Foundation.
SAN DIEGO – In patients with early rheumatoid arthritis, treatment with a combination of disease-modifying antirheumatic drugs is more cost effective than an immediate or step-up approach using etanercept and methotrexate, a new analysis demonstrated.
"The benefits from all strategies were comparable, but biologics strategies were almost twice as expensive [as] triple strategies, producing incremental cost-effectiveness ratios higher than what most health care settings would find acceptable," Kaleb Michaud, Ph.D., said at the annual meeting of the American College of Rheumatology.
Dr. Michaud of the division of rheumatology and immunology at the University of Nebraska Medical Center, Omaha, and his associates analyzed data from 755 patients enrolled in the randomized, 2-year TEAR (Treatment of Early Aggressive RA) trial (Arthritis Rheum. 2012;64:2824-35), which compared the long-term effectiveness of using a triple therapy approach for treating RA with a strategy of using a etanercept (Enbrel) plus methotrexate. They studied four approaches: immediate triple therapy of methotrexate, sulfasalazine, and hydroxychloroquine; immediate treatment with etanercept and methotrexate; a step-up triple therapy; and a step-up etanercept therapy. The researchers incorporated estimates of annual discontinuation rates of triple therapy and etanercept of 22% and 10%, respectively, based on data from the National Data Bank for Rheumatic Diseases.
The analysis was limited to patients with RA duration of less than 3 years and those with active disease, defined as having four or more swollen joints with seropositivity and/or erosions. The mean disease duration was 3.6 months and the mean Disease Activity Score was 5.8. The researchers used a societal perspective and a Markov cohort model with 3% discounting to estimate quality-adjusted life year (QALY) measurements and the costs associated with therapy approaches in the TEAR trial.
Dr. Michaud, who is also codirector of the National Data Bank for Rheumatic Diseases, reported that the lifetime benefit of all four treatment approaches were comparable, within 0.06 QALYs of each other. However, the therapies that included etanercept were nearly twice as expensive because of the higher cost of the TNF inhibitor. Specifically, the costs were $152,400 in the immediate triple therapy group and $154,900 in the step-up triple therapy group, compared with $269,500 in the step-up etanercept group and $338,100 in the immediate etanercept group. The researchers determined that the lifetime incremental cost-effectiveness ratio of immediate etanercept to immediate triple therapy was $837,100/QALY.
"The immediate triple therapy dominated other arms at 1 and 2 years in this trial, and its results were most sensitive to the costs of etanercept and the reduction in triple therapy discontinuation rates," Dr. Michaud commented. "Our sensitivity analysis did not change our conclusions."
Dr. Michaud disclosed that the study was supported in part by a 2012 investigator award from the Rheumatology Research Foundation.
SAN DIEGO – In patients with early rheumatoid arthritis, treatment with a combination of disease-modifying antirheumatic drugs is more cost effective than an immediate or step-up approach using etanercept and methotrexate, a new analysis demonstrated.
"The benefits from all strategies were comparable, but biologics strategies were almost twice as expensive [as] triple strategies, producing incremental cost-effectiveness ratios higher than what most health care settings would find acceptable," Kaleb Michaud, Ph.D., said at the annual meeting of the American College of Rheumatology.
Dr. Michaud of the division of rheumatology and immunology at the University of Nebraska Medical Center, Omaha, and his associates analyzed data from 755 patients enrolled in the randomized, 2-year TEAR (Treatment of Early Aggressive RA) trial (Arthritis Rheum. 2012;64:2824-35), which compared the long-term effectiveness of using a triple therapy approach for treating RA with a strategy of using a etanercept (Enbrel) plus methotrexate. They studied four approaches: immediate triple therapy of methotrexate, sulfasalazine, and hydroxychloroquine; immediate treatment with etanercept and methotrexate; a step-up triple therapy; and a step-up etanercept therapy. The researchers incorporated estimates of annual discontinuation rates of triple therapy and etanercept of 22% and 10%, respectively, based on data from the National Data Bank for Rheumatic Diseases.
The analysis was limited to patients with RA duration of less than 3 years and those with active disease, defined as having four or more swollen joints with seropositivity and/or erosions. The mean disease duration was 3.6 months and the mean Disease Activity Score was 5.8. The researchers used a societal perspective and a Markov cohort model with 3% discounting to estimate quality-adjusted life year (QALY) measurements and the costs associated with therapy approaches in the TEAR trial.
Dr. Michaud, who is also codirector of the National Data Bank for Rheumatic Diseases, reported that the lifetime benefit of all four treatment approaches were comparable, within 0.06 QALYs of each other. However, the therapies that included etanercept were nearly twice as expensive because of the higher cost of the TNF inhibitor. Specifically, the costs were $152,400 in the immediate triple therapy group and $154,900 in the step-up triple therapy group, compared with $269,500 in the step-up etanercept group and $338,100 in the immediate etanercept group. The researchers determined that the lifetime incremental cost-effectiveness ratio of immediate etanercept to immediate triple therapy was $837,100/QALY.
"The immediate triple therapy dominated other arms at 1 and 2 years in this trial, and its results were most sensitive to the costs of etanercept and the reduction in triple therapy discontinuation rates," Dr. Michaud commented. "Our sensitivity analysis did not change our conclusions."
Dr. Michaud disclosed that the study was supported in part by a 2012 investigator award from the Rheumatology Research Foundation.
AT THE ACR ANNUAL MEETING
Major finding: The incremental cost-effectiveness ratio of immediate etanercept to immediate triple therapy was $837,100/quality-adjusted life year.
Data source: An analysis of 755 patients enrolled in the randomized, 2-year Treatment of Early Aggressive RA (TEAR) trial and patient-level data from the National Data Bank for Rheumatic Diseases.
Disclosures: Dr. Michaud disclosed that the study was supported in part by a 2012 investigator award from the Rheumatology Research Foundation.
Cyclophosphamide and rituximab combo reduced severe lupus flares
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
AT THE ACR ANNUAL MEETING
Major finding: Patients with severe recurrent systemic lupus erythematosus collectively had 38 lupus flares, including 26 with renal involvement, during a median 36-month period before undergoing combination therapy with intravenous cyclophosphamide and rituximab; they had 13 flares, only 3 of which featured renal involvement, during 21 months afterward.
Data source: A retrospective study including 43 patients with severe recurrent SLE, 31 of whom were placed on intravenous cyclophosphamide monotherapy, while 12 received combination therapy with cyclophosphamide plus rituximab.
Disclosures: This study was sponsored by the National Institutes of Health. The presenter, an employee of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, reported having no relevant financial conflicts.
Low serum uric acid levels protect against progressions of renal disease
SAN DIEGO – Patients who achieve a serum uric acid level of less than 6 mg/dL based on current gout management guidelines demonstrated a 37% reduction in progression of renal disease, a large retrospective study showed.
"There are numerous studies showing that people with renal disease can develop hyperuricemia," Dr. Gerald D. Levy said during a press briefing at the annual meeting of the American College of Rheumatology. "Some of them will also develop gout. There are a few small studies showing that in humans, you can reverse hyperuricemia with urate lowering therapy and make an impact in renal disease. We wanted to see if this is true."
Dr. Levy of the division of rheumatology in the department of internal medicine at Kaiser Permanente Medical Group, Downey, Calif., was the lead investigators in a study of 111,992 Kaiser Permanente Southern California patients with a serum uric acid (SUA) level of 7 mg/dL or greater from Jan. 1, 2002, to Dec. 31, 2010. Patients with at least 12 months of health plan membership, including drug benefit prior to the index date, were studied. All patients had at least one SUA and glomerular filtration rate (GFR) level measurement in the 6-month period prior to the index date and at least one SUA and one GFR in the follow-up period following the index date. Primary outcome events were at least a 30% worsening of renal function, initiation of dialysis, having a GFR of less than 15 mL/min, and undergoing a kidney transplant.
Patients with a new diagnosis of cancer were excluded from the analysis, as were those with HIV, glomerulonephritis, and/or organ transplant other than a kidney transplant.
Dr. Levy reported results from 16,186 patients who were divided into three groups: those who were never treated with urate-lowering therapy (ULT; 11,192); those who were treated with ULT less than 80% of the time from the index date to the end of follow-up period (3,902); and those who were treated with ULT 80% of the time or more from the index date to the end of the follow-up period (1,092). Of the three treatment groups, those who were treated with ULT 80% of the time or more during the study tended to be older and have more comorbid conditions, compared with the other two groups. They also began their ULT therapy earlier.
Among all patients combined, factors significantly associated with renal disease progression included having diabetes (hazard ratio, 1.96), hypertension (HR, 1.50), heart failure (HR, 1.39), previous hospitalizations (HR, 1.33), and being female (HR, 1.49) and older (HR, 1.03). The researchers found that time on ULT was not significantly associated with a reduction in renal disease progression outcome events (HR, 1.27, among those on ULT less than 80% of the time during the study vs. HR, 1.08, among those on ULT 80% of the time or more during the study). However, patients who achieved an SUA level below 6 mg/dL – a treatment goal in the 2012 ACR guidelines for management of gout – demonstrated a 37% reduction in renal disease progression (HR, 0.63; P less than .0001).
Dr. Levy acknowledged certain limitations of the study, including its retrospective design and the fact that patients with stage 4 and 5 chronic kidney disease were not included. "This is an important area, because if we can delay the worsening of renal disease in these folks, perhaps we’re abetting dialysis, which is growing by leaps and bounds in this country," he said. "Each dialysis patient now costs about $80,000 per year to take care of. If we could push that back even for a few years it would have a tremendous impact."
Dr. Levy had no relevant financial conflicts to disclose.
SAN DIEGO – Patients who achieve a serum uric acid level of less than 6 mg/dL based on current gout management guidelines demonstrated a 37% reduction in progression of renal disease, a large retrospective study showed.
"There are numerous studies showing that people with renal disease can develop hyperuricemia," Dr. Gerald D. Levy said during a press briefing at the annual meeting of the American College of Rheumatology. "Some of them will also develop gout. There are a few small studies showing that in humans, you can reverse hyperuricemia with urate lowering therapy and make an impact in renal disease. We wanted to see if this is true."
Dr. Levy of the division of rheumatology in the department of internal medicine at Kaiser Permanente Medical Group, Downey, Calif., was the lead investigators in a study of 111,992 Kaiser Permanente Southern California patients with a serum uric acid (SUA) level of 7 mg/dL or greater from Jan. 1, 2002, to Dec. 31, 2010. Patients with at least 12 months of health plan membership, including drug benefit prior to the index date, were studied. All patients had at least one SUA and glomerular filtration rate (GFR) level measurement in the 6-month period prior to the index date and at least one SUA and one GFR in the follow-up period following the index date. Primary outcome events were at least a 30% worsening of renal function, initiation of dialysis, having a GFR of less than 15 mL/min, and undergoing a kidney transplant.
Patients with a new diagnosis of cancer were excluded from the analysis, as were those with HIV, glomerulonephritis, and/or organ transplant other than a kidney transplant.
Dr. Levy reported results from 16,186 patients who were divided into three groups: those who were never treated with urate-lowering therapy (ULT; 11,192); those who were treated with ULT less than 80% of the time from the index date to the end of follow-up period (3,902); and those who were treated with ULT 80% of the time or more from the index date to the end of the follow-up period (1,092). Of the three treatment groups, those who were treated with ULT 80% of the time or more during the study tended to be older and have more comorbid conditions, compared with the other two groups. They also began their ULT therapy earlier.
Among all patients combined, factors significantly associated with renal disease progression included having diabetes (hazard ratio, 1.96), hypertension (HR, 1.50), heart failure (HR, 1.39), previous hospitalizations (HR, 1.33), and being female (HR, 1.49) and older (HR, 1.03). The researchers found that time on ULT was not significantly associated with a reduction in renal disease progression outcome events (HR, 1.27, among those on ULT less than 80% of the time during the study vs. HR, 1.08, among those on ULT 80% of the time or more during the study). However, patients who achieved an SUA level below 6 mg/dL – a treatment goal in the 2012 ACR guidelines for management of gout – demonstrated a 37% reduction in renal disease progression (HR, 0.63; P less than .0001).
Dr. Levy acknowledged certain limitations of the study, including its retrospective design and the fact that patients with stage 4 and 5 chronic kidney disease were not included. "This is an important area, because if we can delay the worsening of renal disease in these folks, perhaps we’re abetting dialysis, which is growing by leaps and bounds in this country," he said. "Each dialysis patient now costs about $80,000 per year to take care of. If we could push that back even for a few years it would have a tremendous impact."
Dr. Levy had no relevant financial conflicts to disclose.
SAN DIEGO – Patients who achieve a serum uric acid level of less than 6 mg/dL based on current gout management guidelines demonstrated a 37% reduction in progression of renal disease, a large retrospective study showed.
"There are numerous studies showing that people with renal disease can develop hyperuricemia," Dr. Gerald D. Levy said during a press briefing at the annual meeting of the American College of Rheumatology. "Some of them will also develop gout. There are a few small studies showing that in humans, you can reverse hyperuricemia with urate lowering therapy and make an impact in renal disease. We wanted to see if this is true."
Dr. Levy of the division of rheumatology in the department of internal medicine at Kaiser Permanente Medical Group, Downey, Calif., was the lead investigators in a study of 111,992 Kaiser Permanente Southern California patients with a serum uric acid (SUA) level of 7 mg/dL or greater from Jan. 1, 2002, to Dec. 31, 2010. Patients with at least 12 months of health plan membership, including drug benefit prior to the index date, were studied. All patients had at least one SUA and glomerular filtration rate (GFR) level measurement in the 6-month period prior to the index date and at least one SUA and one GFR in the follow-up period following the index date. Primary outcome events were at least a 30% worsening of renal function, initiation of dialysis, having a GFR of less than 15 mL/min, and undergoing a kidney transplant.
Patients with a new diagnosis of cancer were excluded from the analysis, as were those with HIV, glomerulonephritis, and/or organ transplant other than a kidney transplant.
Dr. Levy reported results from 16,186 patients who were divided into three groups: those who were never treated with urate-lowering therapy (ULT; 11,192); those who were treated with ULT less than 80% of the time from the index date to the end of follow-up period (3,902); and those who were treated with ULT 80% of the time or more from the index date to the end of the follow-up period (1,092). Of the three treatment groups, those who were treated with ULT 80% of the time or more during the study tended to be older and have more comorbid conditions, compared with the other two groups. They also began their ULT therapy earlier.
Among all patients combined, factors significantly associated with renal disease progression included having diabetes (hazard ratio, 1.96), hypertension (HR, 1.50), heart failure (HR, 1.39), previous hospitalizations (HR, 1.33), and being female (HR, 1.49) and older (HR, 1.03). The researchers found that time on ULT was not significantly associated with a reduction in renal disease progression outcome events (HR, 1.27, among those on ULT less than 80% of the time during the study vs. HR, 1.08, among those on ULT 80% of the time or more during the study). However, patients who achieved an SUA level below 6 mg/dL – a treatment goal in the 2012 ACR guidelines for management of gout – demonstrated a 37% reduction in renal disease progression (HR, 0.63; P less than .0001).
Dr. Levy acknowledged certain limitations of the study, including its retrospective design and the fact that patients with stage 4 and 5 chronic kidney disease were not included. "This is an important area, because if we can delay the worsening of renal disease in these folks, perhaps we’re abetting dialysis, which is growing by leaps and bounds in this country," he said. "Each dialysis patient now costs about $80,000 per year to take care of. If we could push that back even for a few years it would have a tremendous impact."
Dr. Levy had no relevant financial conflicts to disclose.
AT THE ACR ANNUAL MEETING
Major finding: Patients who achieved a serum uric acid level below 6 mg/dL – a treatment goal in the 2012 ACR guidelines for management of gout – demonstrated a 37% reduction in renal disease progression (HR, 0.63; P less than .0001).
Data source: A study of 16,186 patients who were divided into three groups: those who were never treated with urate-lowering therapy (ULT; 11,192), those who were treated with ULT less than 80% of the time from the index date to the end of follow-up period (3,902), and those who were treated with ULT 80% of the time or more from the index date to the end of the follow-up period (1,092).
Disclosures: Dr. Levy said that he had no relevant financial conflicts to disclose.

Delayed-release prednisone lessened rheumatoid arthritis symptoms
SAN DIEGO – Rheumatoid arthritis patients who switched from conventional immediate-release prednisone to delayed-release prednisone taken at bedtime experienced a mean reduction of more than 60 minutes in daily morning stiffness that was maintained throughout a 9-month open-label study.
This reduction from a mean baseline of 143.5 minutes of daily morning stiffness during 3 months on a mean 6.8 mg/day of immediate-release prednisone to a similar dose of proprietary delayed-release prednisone (Rayos) constitutes a clinically meaningful benefit. It was accompanied by improvements in other important patient-reported outcomes, Dr. Allan Gibofsky observed at the annual meeting of the American College of Rheumatology.
He presented an analysis of the 9-month open-label phase of the CAPRA-1 (Circadian Administration of Prednisone in Rheumatoid Arthritis-1) study, which also featured a previously reported double-blind phase. The open-label phase involved 207 patients with baseline moderate-to-severe active RA on a stable regimen of disease-modifying antirheumatic drug (DMARD) therapy. After 3 months on immediate-release prednisone during the double-blind phase of CAPRI-1, 110 study participants were switched to delayed-release prednisone; the other 97 had been on delayed-release prednisone during the double-blind phase and continued taking it at about 10 p.m. during the open-label phase.
Levels of the inflammatory cytokine interleukin-6 (IL-6) were measured at the start and conclusion of the 9-month open-label study. The 107 patients who switched from immediate- to delayed-release prednisone showed a median 53% drop in IL-6 levels from a baseline of 1,055 IU/L, reported Dr. Gibofsky, professor of medicine and public health at Cornell University and a rheumatologist at the Hospital for Special Surgery, both in New York.
Self-reported pain scores on a 0-100 visual analog scale dropped from a baseline of 44 by a mean 6.1 points after the switch to delayed-release prednisone and remained stable thereafter. In addition, patient-reported global assessment improved significantly at 3 and 6 months despite 3 months of previous double-blind therapy with immediate-release prednisone.
Delayed-release prednisone is approved as an anti-inflammatory or immunosuppressive agent for a wide array of dermatologic, respiratory, GI, renal, and rheumatologic disorders, including RA. The underlying rationale for this form of chronotherapy is that it permits timed delivery of prednisone during the early morning hours in accord with the circadian pattern of inflammatory cytokine levels.
CAPRI-1 was funded by Horizon Pharma, which markets Rayos. Dr. Gibofsky is a consultant to the company.
SAN DIEGO – Rheumatoid arthritis patients who switched from conventional immediate-release prednisone to delayed-release prednisone taken at bedtime experienced a mean reduction of more than 60 minutes in daily morning stiffness that was maintained throughout a 9-month open-label study.
This reduction from a mean baseline of 143.5 minutes of daily morning stiffness during 3 months on a mean 6.8 mg/day of immediate-release prednisone to a similar dose of proprietary delayed-release prednisone (Rayos) constitutes a clinically meaningful benefit. It was accompanied by improvements in other important patient-reported outcomes, Dr. Allan Gibofsky observed at the annual meeting of the American College of Rheumatology.
He presented an analysis of the 9-month open-label phase of the CAPRA-1 (Circadian Administration of Prednisone in Rheumatoid Arthritis-1) study, which also featured a previously reported double-blind phase. The open-label phase involved 207 patients with baseline moderate-to-severe active RA on a stable regimen of disease-modifying antirheumatic drug (DMARD) therapy. After 3 months on immediate-release prednisone during the double-blind phase of CAPRI-1, 110 study participants were switched to delayed-release prednisone; the other 97 had been on delayed-release prednisone during the double-blind phase and continued taking it at about 10 p.m. during the open-label phase.
Levels of the inflammatory cytokine interleukin-6 (IL-6) were measured at the start and conclusion of the 9-month open-label study. The 107 patients who switched from immediate- to delayed-release prednisone showed a median 53% drop in IL-6 levels from a baseline of 1,055 IU/L, reported Dr. Gibofsky, professor of medicine and public health at Cornell University and a rheumatologist at the Hospital for Special Surgery, both in New York.
Self-reported pain scores on a 0-100 visual analog scale dropped from a baseline of 44 by a mean 6.1 points after the switch to delayed-release prednisone and remained stable thereafter. In addition, patient-reported global assessment improved significantly at 3 and 6 months despite 3 months of previous double-blind therapy with immediate-release prednisone.
Delayed-release prednisone is approved as an anti-inflammatory or immunosuppressive agent for a wide array of dermatologic, respiratory, GI, renal, and rheumatologic disorders, including RA. The underlying rationale for this form of chronotherapy is that it permits timed delivery of prednisone during the early morning hours in accord with the circadian pattern of inflammatory cytokine levels.
CAPRI-1 was funded by Horizon Pharma, which markets Rayos. Dr. Gibofsky is a consultant to the company.
SAN DIEGO – Rheumatoid arthritis patients who switched from conventional immediate-release prednisone to delayed-release prednisone taken at bedtime experienced a mean reduction of more than 60 minutes in daily morning stiffness that was maintained throughout a 9-month open-label study.
This reduction from a mean baseline of 143.5 minutes of daily morning stiffness during 3 months on a mean 6.8 mg/day of immediate-release prednisone to a similar dose of proprietary delayed-release prednisone (Rayos) constitutes a clinically meaningful benefit. It was accompanied by improvements in other important patient-reported outcomes, Dr. Allan Gibofsky observed at the annual meeting of the American College of Rheumatology.
He presented an analysis of the 9-month open-label phase of the CAPRA-1 (Circadian Administration of Prednisone in Rheumatoid Arthritis-1) study, which also featured a previously reported double-blind phase. The open-label phase involved 207 patients with baseline moderate-to-severe active RA on a stable regimen of disease-modifying antirheumatic drug (DMARD) therapy. After 3 months on immediate-release prednisone during the double-blind phase of CAPRI-1, 110 study participants were switched to delayed-release prednisone; the other 97 had been on delayed-release prednisone during the double-blind phase and continued taking it at about 10 p.m. during the open-label phase.
Levels of the inflammatory cytokine interleukin-6 (IL-6) were measured at the start and conclusion of the 9-month open-label study. The 107 patients who switched from immediate- to delayed-release prednisone showed a median 53% drop in IL-6 levels from a baseline of 1,055 IU/L, reported Dr. Gibofsky, professor of medicine and public health at Cornell University and a rheumatologist at the Hospital for Special Surgery, both in New York.
Self-reported pain scores on a 0-100 visual analog scale dropped from a baseline of 44 by a mean 6.1 points after the switch to delayed-release prednisone and remained stable thereafter. In addition, patient-reported global assessment improved significantly at 3 and 6 months despite 3 months of previous double-blind therapy with immediate-release prednisone.
Delayed-release prednisone is approved as an anti-inflammatory or immunosuppressive agent for a wide array of dermatologic, respiratory, GI, renal, and rheumatologic disorders, including RA. The underlying rationale for this form of chronotherapy is that it permits timed delivery of prednisone during the early morning hours in accord with the circadian pattern of inflammatory cytokine levels.
CAPRI-1 was funded by Horizon Pharma, which markets Rayos. Dr. Gibofsky is a consultant to the company.
AT THE ACR ANNUAL MEETING
Major finding: Patients with moderate to severe active rheumatoid arthritis on stable disease-modifying antirheumatic drug therapy experienced a greater than 60-minute reduction in daily morning stiffness from a baseline of 143.5 minutes when they were switched from immediate- to delayed-release prednisone.
Data source: This was an analysis of the 207 RA patients who participated in the 9-month open-label phase of the prospective CAPRA-1 study.
Disclosures: The study was funded by Horizon Pharma, which markets immediate-release prednisone. Dr. Gibofsky is a consultant to the company.
Unsuspected Primary Hyperparathyroidism Common in Fibromyalgia Patients
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
AT THE ACR ANNUAL MEETING
Unsuspected primary hyperparathyroidism common in fibromyalgia patients
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
SAN DIEGO – The prevalence of unsuspected primary hyperparathyroidism among patients diagnosed with fibromyalgia was 11% in a small exploratory study, Dr. Michael Tsoukas reported at the annual meeting of the American College of Rheumatology.
That’s roughly 100-fold greater than the prevalence of primary hyperparathyroidism in the general population, noted Dr. Tsoukas of McGill University, Montreal.
The classic symptoms of primary hyperparathyroidism include musculoskeletal pain, fatigue, mood disorders, and sleep disturbances, a clinical picture closely mimicking that of fibromyalgia, he observed.
Dr. Tsoukas presented a retrospective study of a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain center where physicians obtained routine blood tests. Four of the 38, or 11%, met criteria for the biochemical diagnosis of hyperthyroidism. Two had what’s known as normohormonal hyperparathyroidism, marked by a serum ionized calcium elevated above 1.32 mmol/L or 5.3 mg/dL, with inappropriately high parathyroid hormone levels. The other two had a parathyroid hormone level in excess of 9.3 pmol/L or 93 pg/mL, with inappropriately nonsuppressed calcium.
This was a pilot study. Four cases of unsuspected hyperparathyroidism hardly make for a definitive study. But this was the first study to look at hyperparathyroidism in fibromyalgia patients, and given how common fibromyalgia is and the difficulties and frustrations often encountered in its treatment, these intriguing preliminary findings warrant large, prospective, multicenter studies to further clarify the relationship between these two disorders with closely similar symptoms, Dr. Tsoukas concluded.
He reported having no financial conflicts of interest in this unfunded study.
AT THE ACR ANNUAL MEETING
Major finding: Four of 38 patients with a primary diagnosis of fibromyalgia proved to have primary hyperparathyroidism. That 11% prevalence is roughly 100 times greater than in the general population.
Data source: This was a chart review involving a convenience sample of 38 consecutive patients with a primary diagnosis of fibromyalgia attending a multidisciplinary tertiary care pain clinic.
Disclosures: Dr. Tsoukas reported having no financial conflicts of interest in this unfunded study.
Encouraging results for oral JAK 3 inhibitor in rheumatoid arthritis
SAN DIEGO – A novel oral Janus kinase 3 inhibitor known for now as VX-509 readily hit both of its coprimary endpoints in a phase IIb study in rheumatoid arthritis patients presented at the annual meeting of the American College of Rheumatology.
The 24-week, double-blind, international study included 358 patients with active rheumatoid arthritis on stable doses of background methotrexate who were randomized to one of four VX-509 dosing regimens or placebo.
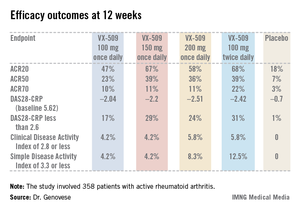
An ACR20 response at 12 weeks occurred in 47%-68% of subjects on various doses of the oral JAK 3 inhibitor, compared with 18% of controls. Robust improvements in the Disease Activity Score 28 C-reactive protein (DAS28-CRP) were also noted in the VX-509-treated patients. The improvement in these two primary endpoints was rapid, with the oral JAK 3 inhibitor’s advantage over placebo becoming significant during the first week, reported Dr. Mark C. Genovese, professor of medicine at Stanford (Calif.) University.
Infections, the most common type of adverse events, occurred in 22% of patients on VX-509 and 15.5% on placebo. Serious infections occurred in 2.8% of patients on the JAK 3 inhibitor, twice the rate observed in controls. Bronchitis, pneumonia, cellulitis, and one severe case of herpes zoster accounted for most of the serious infections in the VX-509 group.

Modest elevations in transaminase levels and reductions in median neutrophil and lymphocyte counts occurred in the VX-509 groups. In addition, dose-dependent increases were observed in low-density lipoprotein (LDL) cholesterol and fasting triglycerides. The LDL increases ranged from 3.1 to 11.6 mg/dL, while triglyceride levels in VX-509–treated patients climbed by 25.7-39 mg/dL.
JAK 3 is exclusively involved in immune function and prevents signaling by inflammatory cytokines, including interleukin-2, -4, -7, -9, -15, and -21. VX-509’s high degree of selectivity for JAK 3 is desirable, Dr. Genovese explained, because the drug doesn’t target JAK 2, which is involved growth factor and hematopoietic signaling and whose inhibition could thereby lead to unwanted effects.
Vertex Pharmaceuticals, which sponsored the phase IIb study, subsequently announced its strong interest in further developing VX-509 for the marketplace. Dr. Genovese disclosed having received research grants from and serving as a consultant to Vertex.
SAN DIEGO – A novel oral Janus kinase 3 inhibitor known for now as VX-509 readily hit both of its coprimary endpoints in a phase IIb study in rheumatoid arthritis patients presented at the annual meeting of the American College of Rheumatology.
The 24-week, double-blind, international study included 358 patients with active rheumatoid arthritis on stable doses of background methotrexate who were randomized to one of four VX-509 dosing regimens or placebo.
An ACR20 response at 12 weeks occurred in 47%-68% of subjects on various doses of the oral JAK 3 inhibitor, compared with 18% of controls. Robust improvements in the Disease Activity Score 28 C-reactive protein (DAS28-CRP) were also noted in the VX-509-treated patients. The improvement in these two primary endpoints was rapid, with the oral JAK 3 inhibitor’s advantage over placebo becoming significant during the first week, reported Dr. Mark C. Genovese, professor of medicine at Stanford (Calif.) University.
Infections, the most common type of adverse events, occurred in 22% of patients on VX-509 and 15.5% on placebo. Serious infections occurred in 2.8% of patients on the JAK 3 inhibitor, twice the rate observed in controls. Bronchitis, pneumonia, cellulitis, and one severe case of herpes zoster accounted for most of the serious infections in the VX-509 group.
Modest elevations in transaminase levels and reductions in median neutrophil and lymphocyte counts occurred in the VX-509 groups. In addition, dose-dependent increases were observed in low-density lipoprotein (LDL) cholesterol and fasting triglycerides. The LDL increases ranged from 3.1 to 11.6 mg/dL, while triglyceride levels in VX-509–treated patients climbed by 25.7-39 mg/dL.
JAK 3 is exclusively involved in immune function and prevents signaling by inflammatory cytokines, including interleukin-2, -4, -7, -9, -15, and -21. VX-509’s high degree of selectivity for JAK 3 is desirable, Dr. Genovese explained, because the drug doesn’t target JAK 2, which is involved growth factor and hematopoietic signaling and whose inhibition could thereby lead to unwanted effects.
Vertex Pharmaceuticals, which sponsored the phase IIb study, subsequently announced its strong interest in further developing VX-509 for the marketplace. Dr. Genovese disclosed having received research grants from and serving as a consultant to Vertex.
SAN DIEGO – A novel oral Janus kinase 3 inhibitor known for now as VX-509 readily hit both of its coprimary endpoints in a phase IIb study in rheumatoid arthritis patients presented at the annual meeting of the American College of Rheumatology.
The 24-week, double-blind, international study included 358 patients with active rheumatoid arthritis on stable doses of background methotrexate who were randomized to one of four VX-509 dosing regimens or placebo.
An ACR20 response at 12 weeks occurred in 47%-68% of subjects on various doses of the oral JAK 3 inhibitor, compared with 18% of controls. Robust improvements in the Disease Activity Score 28 C-reactive protein (DAS28-CRP) were also noted in the VX-509-treated patients. The improvement in these two primary endpoints was rapid, with the oral JAK 3 inhibitor’s advantage over placebo becoming significant during the first week, reported Dr. Mark C. Genovese, professor of medicine at Stanford (Calif.) University.
Infections, the most common type of adverse events, occurred in 22% of patients on VX-509 and 15.5% on placebo. Serious infections occurred in 2.8% of patients on the JAK 3 inhibitor, twice the rate observed in controls. Bronchitis, pneumonia, cellulitis, and one severe case of herpes zoster accounted for most of the serious infections in the VX-509 group.
Modest elevations in transaminase levels and reductions in median neutrophil and lymphocyte counts occurred in the VX-509 groups. In addition, dose-dependent increases were observed in low-density lipoprotein (LDL) cholesterol and fasting triglycerides. The LDL increases ranged from 3.1 to 11.6 mg/dL, while triglyceride levels in VX-509–treated patients climbed by 25.7-39 mg/dL.
JAK 3 is exclusively involved in immune function and prevents signaling by inflammatory cytokines, including interleukin-2, -4, -7, -9, -15, and -21. VX-509’s high degree of selectivity for JAK 3 is desirable, Dr. Genovese explained, because the drug doesn’t target JAK 2, which is involved growth factor and hematopoietic signaling and whose inhibition could thereby lead to unwanted effects.
Vertex Pharmaceuticals, which sponsored the phase IIb study, subsequently announced its strong interest in further developing VX-509 for the marketplace. Dr. Genovese disclosed having received research grants from and serving as a consultant to Vertex.
AT THE ACR ANNUAL MEETING
Major finding: Rheumatoid arthritis patients had ACR20 responses of 47%-68% to various dosing regimens of the novel oral Janus kinase 3 inhibitor VX-509 at 12 weeks, compared with 18% in placebo-treated controls.
Data source: This was a phase IIb, double-blind, placebo-controlled, international study involving 358 patients with rheumatoid arthritis on background stable doses of methotrexate, continued during the trial.
Disclosures: The study was sponsored by Vertex Pharmaceuticals. The presenter disclosed receiving research grants from and serving as a consultant to Vertex.
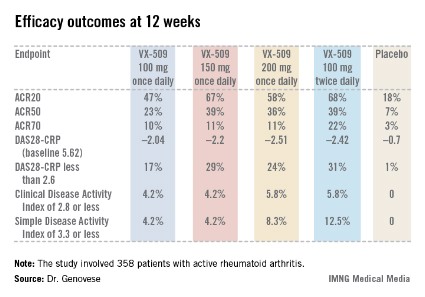
Many gout patients not reaching treatment goals
SAN DIEGO – A high percentage of gout patients treated by rheumatologists do not meet the treatment goals established by the American College of Rheumatology, even after 6 months of higher-dose urate-lowering therapy, results from a national survey found.
"These findings suggest that even among rheumatologists, gout management may not be optimal and may be inadequately aggressive in the most severe patients," Dr. Max Hamburger said at the annual meeting of the American College of Rheumatology. "It seems that there is further study needed to determine the long-term impact of the new ACR guidelines."
In 2012 the ACR published updated guidelines for the management of gout and hyperuricemia (Arth. Care and Res. 2012;64:1431-46). The recommendations included a call for treat-to-target serum uric acid (sUA) of below 6 mg/dL at a minimum, and below 5 mg/dL in select patients. "The intent of this treat-to-target was to durably improve signs and symptoms of gout and also to address palpable and visible tophi," said Dr. Hamburger, a rheumatologist who practices in Melville, N.Y. "The extent to which current practice among rheumatologists aligns with the guidelines is unknown. The areas in which the guidelines may help improve gout treatment also remains to be determined."

He and his associates set out to assess symptoms, treatment, and outcomes among gout patients treated by rheumatologists in the United States and to identify gaps that might exist in current practice with the new ACR recommendations. They recruited a national sample of rheumatologists to report gout patient encounters prospectively during Jan. 15 to Feb. 22, 2013. Rheumatologists were eligible for the study if they were board certified or board eligible in rheumatology, if they spent at least 70% of their time on patient care, if they were in practice for at least 2 years, and if they saw at least four gout patients per month.
The researchers collected anonymous patient data, including demographics, history with the rheumatology practice, gout symptoms and severity, rheumatologist assessment of disease control, and gout medications and treatment changes at the time of each visit. They applied the ACR working case scenarios and grouped patients by increasing level of disease severity. Patients in the scenarios 1-3 group had intermittent symptoms and no tophi (mild disease); patients in the scenarios 4-6 group had intermittent symptoms and 1 tophus or more (moderate disease), and patients in the scenarios 7-9 group had chronic tophaceous gouty arthropathy (more severe disease). Higher-dose ULT was defined as greater than 300 mg/day of allopurinol or 80 mg or more per day of febuxostat (Uloric).
Dr. Hamburger reported results from 127 rheumatologists who received 2,380 valid patient encounter forms. Most of the patients (79%) were male, their mean age was 61 years, and 72% were seen by a rheumatologist for 6 months or longer. Based on ACR scenario groupings, 68% were in the scenarios 1-3 group, 4% were in the scenarios 4-6 group, and 28% were in the scenarios 7-9 group.
Most patients in the scenarios 1-3 group were judged by the rheumatologists to have controlled disease, compared with 91% of patients in the scenarios 4-6 group and 81% of patients in the scenarios 7-9 group. In addition, 14% of patients in the scenarios 1-3 group were on higher-dose ULT, compared with 28% in the scenarios 4-6 group and 40% in the scenarios 7-9 group. Nearly one-quarter of all patients (24%) were on higher-dose ULT.
Among patients on higher-dose ULT, 45% of those in the scenarios 1-3 group had an sUA greater than 6 mg/dL, compared with 53% in the scenarios 4-6 group and 61% in the scenarios 7-9 group. "Despite elevated sUA, 45% of encounters did not result in an increased ULT dose or treatment change at this visit," Dr. Hamburger said.
Even with 6 months or more at higher-dose ULT, only 55% of patients overall had an sUA at or below guideline recommendations of 6 mg/dL, including only 40% of patients in the scenarios 7-9 group. In addition, 16% of patients overall had an sUA between 6 and 6.8 mg/dL, despite being on higher-dose ULT for 6 months or longer.
Dr. Hamburger acknowledged certain limitations of the study, including the fact that "rheumatologist participation in this market research may be biased based on willingness to participate in online data collection over the reporting period," he said. In addition, "a varying number of encounter forms were provided by each participant and based on estimated patient volume. Not all participants reported on 100% of their patients during the reporting period."
Dr. Hamburger disclosed that he is a speaker for Savient Pharmaceuticals, which markets the gout drug pegloticase (Krystexxa), and Takeda Pharmaceuticals, which markets febuxostat and colchicine (Colcrys). Funding for the market research used in the study was provided by Savient.
SAN DIEGO – A high percentage of gout patients treated by rheumatologists do not meet the treatment goals established by the American College of Rheumatology, even after 6 months of higher-dose urate-lowering therapy, results from a national survey found.
"These findings suggest that even among rheumatologists, gout management may not be optimal and may be inadequately aggressive in the most severe patients," Dr. Max Hamburger said at the annual meeting of the American College of Rheumatology. "It seems that there is further study needed to determine the long-term impact of the new ACR guidelines."
In 2012 the ACR published updated guidelines for the management of gout and hyperuricemia (Arth. Care and Res. 2012;64:1431-46). The recommendations included a call for treat-to-target serum uric acid (sUA) of below 6 mg/dL at a minimum, and below 5 mg/dL in select patients. "The intent of this treat-to-target was to durably improve signs and symptoms of gout and also to address palpable and visible tophi," said Dr. Hamburger, a rheumatologist who practices in Melville, N.Y. "The extent to which current practice among rheumatologists aligns with the guidelines is unknown. The areas in which the guidelines may help improve gout treatment also remains to be determined."
He and his associates set out to assess symptoms, treatment, and outcomes among gout patients treated by rheumatologists in the United States and to identify gaps that might exist in current practice with the new ACR recommendations. They recruited a national sample of rheumatologists to report gout patient encounters prospectively during Jan. 15 to Feb. 22, 2013. Rheumatologists were eligible for the study if they were board certified or board eligible in rheumatology, if they spent at least 70% of their time on patient care, if they were in practice for at least 2 years, and if they saw at least four gout patients per month.
The researchers collected anonymous patient data, including demographics, history with the rheumatology practice, gout symptoms and severity, rheumatologist assessment of disease control, and gout medications and treatment changes at the time of each visit. They applied the ACR working case scenarios and grouped patients by increasing level of disease severity. Patients in the scenarios 1-3 group had intermittent symptoms and no tophi (mild disease); patients in the scenarios 4-6 group had intermittent symptoms and 1 tophus or more (moderate disease), and patients in the scenarios 7-9 group had chronic tophaceous gouty arthropathy (more severe disease). Higher-dose ULT was defined as greater than 300 mg/day of allopurinol or 80 mg or more per day of febuxostat (Uloric).
Dr. Hamburger reported results from 127 rheumatologists who received 2,380 valid patient encounter forms. Most of the patients (79%) were male, their mean age was 61 years, and 72% were seen by a rheumatologist for 6 months or longer. Based on ACR scenario groupings, 68% were in the scenarios 1-3 group, 4% were in the scenarios 4-6 group, and 28% were in the scenarios 7-9 group.
Most patients in the scenarios 1-3 group were judged by the rheumatologists to have controlled disease, compared with 91% of patients in the scenarios 4-6 group and 81% of patients in the scenarios 7-9 group. In addition, 14% of patients in the scenarios 1-3 group were on higher-dose ULT, compared with 28% in the scenarios 4-6 group and 40% in the scenarios 7-9 group. Nearly one-quarter of all patients (24%) were on higher-dose ULT.
Among patients on higher-dose ULT, 45% of those in the scenarios 1-3 group had an sUA greater than 6 mg/dL, compared with 53% in the scenarios 4-6 group and 61% in the scenarios 7-9 group. "Despite elevated sUA, 45% of encounters did not result in an increased ULT dose or treatment change at this visit," Dr. Hamburger said.
Even with 6 months or more at higher-dose ULT, only 55% of patients overall had an sUA at or below guideline recommendations of 6 mg/dL, including only 40% of patients in the scenarios 7-9 group. In addition, 16% of patients overall had an sUA between 6 and 6.8 mg/dL, despite being on higher-dose ULT for 6 months or longer.
Dr. Hamburger acknowledged certain limitations of the study, including the fact that "rheumatologist participation in this market research may be biased based on willingness to participate in online data collection over the reporting period," he said. In addition, "a varying number of encounter forms were provided by each participant and based on estimated patient volume. Not all participants reported on 100% of their patients during the reporting period."
Dr. Hamburger disclosed that he is a speaker for Savient Pharmaceuticals, which markets the gout drug pegloticase (Krystexxa), and Takeda Pharmaceuticals, which markets febuxostat and colchicine (Colcrys). Funding for the market research used in the study was provided by Savient.
SAN DIEGO – A high percentage of gout patients treated by rheumatologists do not meet the treatment goals established by the American College of Rheumatology, even after 6 months of higher-dose urate-lowering therapy, results from a national survey found.
"These findings suggest that even among rheumatologists, gout management may not be optimal and may be inadequately aggressive in the most severe patients," Dr. Max Hamburger said at the annual meeting of the American College of Rheumatology. "It seems that there is further study needed to determine the long-term impact of the new ACR guidelines."
In 2012 the ACR published updated guidelines for the management of gout and hyperuricemia (Arth. Care and Res. 2012;64:1431-46). The recommendations included a call for treat-to-target serum uric acid (sUA) of below 6 mg/dL at a minimum, and below 5 mg/dL in select patients. "The intent of this treat-to-target was to durably improve signs and symptoms of gout and also to address palpable and visible tophi," said Dr. Hamburger, a rheumatologist who practices in Melville, N.Y. "The extent to which current practice among rheumatologists aligns with the guidelines is unknown. The areas in which the guidelines may help improve gout treatment also remains to be determined."
He and his associates set out to assess symptoms, treatment, and outcomes among gout patients treated by rheumatologists in the United States and to identify gaps that might exist in current practice with the new ACR recommendations. They recruited a national sample of rheumatologists to report gout patient encounters prospectively during Jan. 15 to Feb. 22, 2013. Rheumatologists were eligible for the study if they were board certified or board eligible in rheumatology, if they spent at least 70% of their time on patient care, if they were in practice for at least 2 years, and if they saw at least four gout patients per month.
The researchers collected anonymous patient data, including demographics, history with the rheumatology practice, gout symptoms and severity, rheumatologist assessment of disease control, and gout medications and treatment changes at the time of each visit. They applied the ACR working case scenarios and grouped patients by increasing level of disease severity. Patients in the scenarios 1-3 group had intermittent symptoms and no tophi (mild disease); patients in the scenarios 4-6 group had intermittent symptoms and 1 tophus or more (moderate disease), and patients in the scenarios 7-9 group had chronic tophaceous gouty arthropathy (more severe disease). Higher-dose ULT was defined as greater than 300 mg/day of allopurinol or 80 mg or more per day of febuxostat (Uloric).
Dr. Hamburger reported results from 127 rheumatologists who received 2,380 valid patient encounter forms. Most of the patients (79%) were male, their mean age was 61 years, and 72% were seen by a rheumatologist for 6 months or longer. Based on ACR scenario groupings, 68% were in the scenarios 1-3 group, 4% were in the scenarios 4-6 group, and 28% were in the scenarios 7-9 group.
Most patients in the scenarios 1-3 group were judged by the rheumatologists to have controlled disease, compared with 91% of patients in the scenarios 4-6 group and 81% of patients in the scenarios 7-9 group. In addition, 14% of patients in the scenarios 1-3 group were on higher-dose ULT, compared with 28% in the scenarios 4-6 group and 40% in the scenarios 7-9 group. Nearly one-quarter of all patients (24%) were on higher-dose ULT.
Among patients on higher-dose ULT, 45% of those in the scenarios 1-3 group had an sUA greater than 6 mg/dL, compared with 53% in the scenarios 4-6 group and 61% in the scenarios 7-9 group. "Despite elevated sUA, 45% of encounters did not result in an increased ULT dose or treatment change at this visit," Dr. Hamburger said.
Even with 6 months or more at higher-dose ULT, only 55% of patients overall had an sUA at or below guideline recommendations of 6 mg/dL, including only 40% of patients in the scenarios 7-9 group. In addition, 16% of patients overall had an sUA between 6 and 6.8 mg/dL, despite being on higher-dose ULT for 6 months or longer.
Dr. Hamburger acknowledged certain limitations of the study, including the fact that "rheumatologist participation in this market research may be biased based on willingness to participate in online data collection over the reporting period," he said. In addition, "a varying number of encounter forms were provided by each participant and based on estimated patient volume. Not all participants reported on 100% of their patients during the reporting period."
Dr. Hamburger disclosed that he is a speaker for Savient Pharmaceuticals, which markets the gout drug pegloticase (Krystexxa), and Takeda Pharmaceuticals, which markets febuxostat and colchicine (Colcrys). Funding for the market research used in the study was provided by Savient.
AT THE ACR ANNUAL MEETING
Major finding: Even with 6 months or more at higher-dose urate-lowering therapy, only 55% of gout patients overall had a serum uric acid level at or below ACR guideline recommendations of 6.0 mg/dL, including only 40% of patients with the most severe disease.
Data source: A study of 127 rheumatologists who reported on encounters with 2,380 gout patients during Jan. 15 to Feb. 22, 2013.
Disclosures: Dr. Hamburger disclosed that he is a speaker for Savient Pharmaceuticals and Takeda Pharmaceuticals. Funding for the market research used in the study was provided by Savient.
