Cutaneous polyarteritis nodosa (CPAN) is a rare cutaneous small- to medium-vessel vasculitis of unknown etiology. Clinically it ranges in manifestation from livedo reticularis to large cutaneous ulcers and necrosis. Prognosis is favorable and progression to systemic polyarteritis nodosa is rare. There are multiple treatment options, none of which have proven to be definitively effective. Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibodies, as well as elevated anti–phosphatidylserine-prothrombin complex antibody. These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. We present a case of asymptomatic CPAN and evaluate if treatment should be instituted for asymptomatic disease that presents with abnormal antibody findings.
Cutaneous polyarteritis nodosa should be in the differential of new-onset livedo reticularis.
Workup with biopsy and specific blood work is important.
Treatment options at this time are limited.
References
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
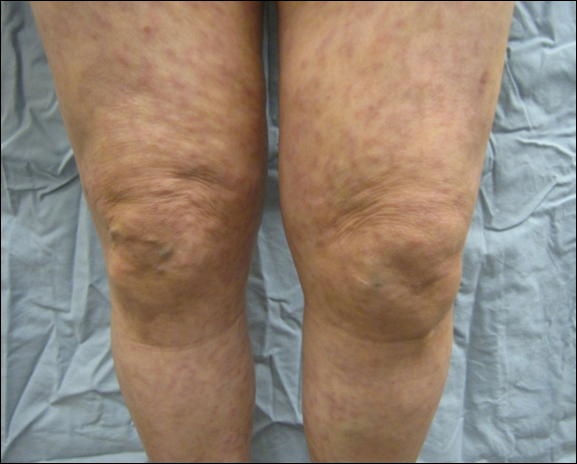
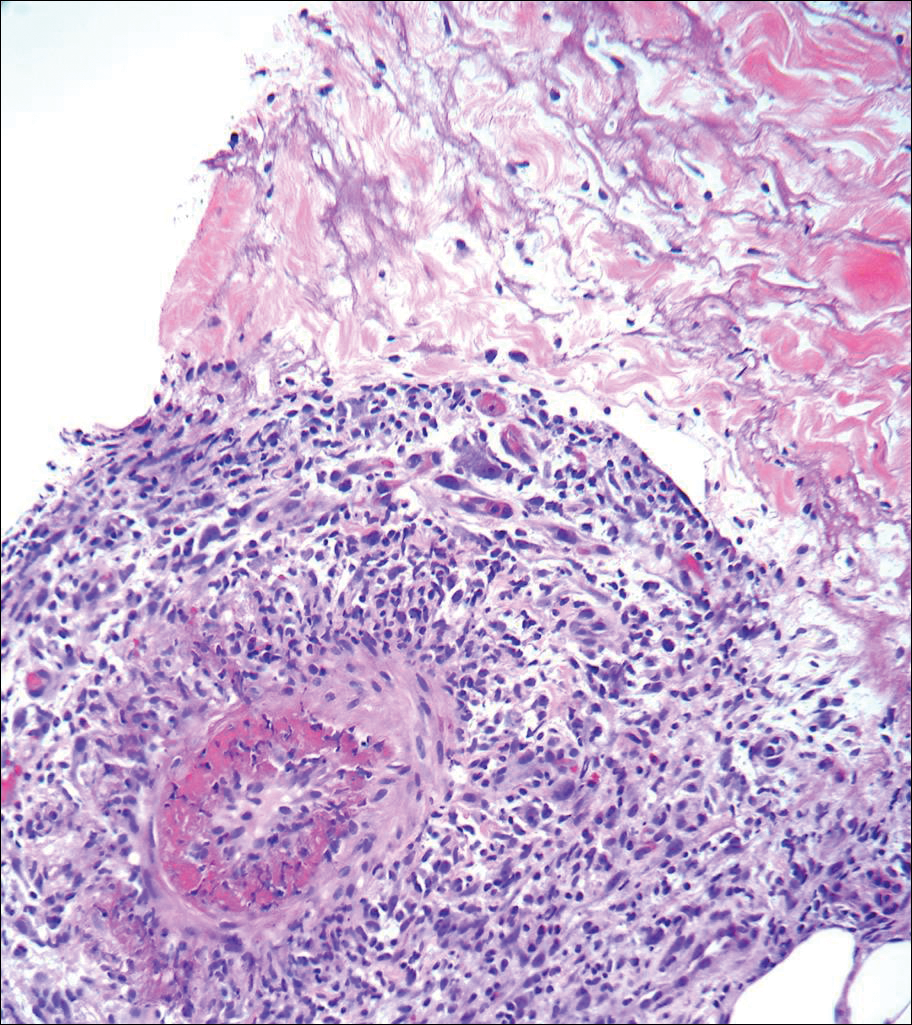
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Figure 1. Livedo reticularis on the legs.
Figure 2. Medium-vessel vasculitis with a lymphocytic infiltrate around a medium-sized vessel (H&E).
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.