User login
Not another burnout article
Does this sound like your day?
You show up to work after a terrible night’s sleep. Your back is tense, and you do some kind of walking/stretching combo as you walk through the doors. Your focus fades during the mind-numbing routine of the morning shift sign out. As the day moves forward, you begin to feel resentful as you sign orders, see patients, and address your ICU team needs. You know that’s not right, that it’s not in line with who you want to be, but the irritation doesn’t go away.
Your lunchtime is filled with computer screens, notes, billing, and more billing. The previous feelings of irritation begin to boil into anger because more of your day is filled with bureaucratic demands and insurance reports rather than actually helping people. This isn’t what you signed up for. Years and years of training so you could be a paper pusher? The thought leads to rage ... or sometimes apathy on days you give in to the inevitable.
You finish your shift with admissions, procedures, code blues, and an overwhelming and exhausting night shift sign out. You feel like a hamster in a wheel. You’re going nowhere. What’s the point of all of this? You find yourself questioning why you went into medicine anyways ... yeah, that’s burnout.
I know what you’re thinking. You keep hearing about this, and it’s important to recognize, but then you hear the same old solutions: be more positive, find balance, do some yoga, take this resilience module, be mindful (what on earth does this mean anyways?), get some more sleep. Basically, it’s our problem. It’s our burden. If all of these were easy to understand and implement, don’t you think doctors and health-care providers would have done it already? I think you and I are a lot alike. These were my exact feelings. But stick with me on this one. I have a solution for you, albeit a little different. I’ll show you a more “positive” spin on the DIY.
I burned out early. After fellowship, I didn’t want to be a doctor anymore. I desperately sought to alter my career somehow. I looked into website development, something I had been good at in high school. I took a few refresher classes on my days off and started coding my own sites, but I had bills to pay. Big bills. Student loan bills. Luckily, my first job out of fellowship accepted many of my schedule demands, such as day shifts only, and after about a year, I recovered and remembered why I had loved medicine to begin with.
What is burnout?
Mind-body-soul exhaustion caused by excessive stress. Stress and burnout are closely related, but they’re more like distant cousins. Stress can be (and is) a normal part of our jobs. I bet you think you’re stressed, when you’re probably burned out. Critical care doctors have the highest rate of burnout among all physician subspecialties at >55%, and it is even higher in pediatric critical care. (Sessler C. https://www.mdedge.com/chestphysician/article/160951/society-news/turning-heat-icu-burnout). The main difference between stress and burnout is hope. With stress, you still feel like things can get better and you can get it all under control. Burnout feels hopeless.
What are the three core symptoms of burnout?
• Irritability and impatience with patients (depersonalization)
• Cynicism and difficulty concentrating (emotional exhaustion)
• What’s the point of all of this? Nothing I do matters or is appreciated (decreased self-efficacy)
We can talk about the symptoms of burnout all day, but what does that really look like? It looks like the day we described at the beginning. You know, the day that resonated with you and caused you to keep reading.
Why should we all be discussing this important topic?
Being burned out not only affects us on a soul level (achingly described above), but, more importantly, this can trickle down to our personal lives, family relationships, and how we care for our patients, with some studies showing that it affects our performance and, gulp, patient outcomes. That’s scary (Moss M et al. Crit Care Med. 2016;44[7]:1414).
Causes of burnout
There are many causes of burnout, and several studies have identified risk factors. A lack of control, conflicts with colleagues and leadership, and performing menial tasks can add to the irritation of a workday. This doesn’t even include the nature of our actual job as critical care doctors. We care for the sickest and are frequently involved in end-of-life care. Over time, the stress morphs into burnout. Female gender is also an independent risk factor for doctors (Pastores SM, et al. Crit Care Med. 2019;47[4]:550).
We’ve identified it. We’ve quantified it. But we’re not fixing it. In fact, there are only a few studies that have incorporated a needs assessment of doctors, paired with appropriate environmental intervention. A study done with primary care doctors in New York City clinics found that surveying a doctor’s “wish list” of interventions can help identify gaps in workflow, such as pairing one medical assistant with each attending (Linzer M, et al. J Gen Intern Med. 2015;30[8]:1105).
Without more data like this, we’re hamsters in a wheel. Luckily, organizations like CHEST have joined together with others to create the Critical Care Societies Collaborative and have an annual summit to discuss research strategies.
Solutions
Even millennials are sick of the mindful “chore” list. Yoga pants, yoga mats, crystals, chakras, meditation, and the list goes on and on. What millennials want are work-life integrations that are easy; workspaces that invite mindful behavior and daily rituals that excite and relax them. Co-working spaces like WeWork have designated self-care spaces.
Self-care is now essential, not an indulgence. I wasn’t sure how to create this space in my ICU, so I started small, with things I could carry with myself. The key is to find small rituals with big meanings. What could this look like for you? I began doing breathwork. Frankly, the idea came to me from my Apple® watch. It just started giving me these reminders one day, and I decided to take it seriously. I found that my mind and muscles eased after only 1 minute of breathing in and out slowly. This elevated my mood and was the refresher I needed in the afternoons. My body ached less after procedures.
I also got a little woo-woo (stay with me now) and began carrying around crystal stones. You don’t have to carry around crystals. Prayer books, religious symbols, your child’s toy car, anything can work if it has meaning for you, so when you see it or touch it during your day, you remember your big why. Why you’re serving people. Why you’re a doctor. I prefer the crystals over jewelry because it’s something unusual that I don’t expect to be sitting in my pocket. It’s always a nice gentle reminder of the love I have for my patients, my job, and humanity. When I put my hands in my pocket as I’m talking to yet another frustrated family member, my responses are more patient and calmer, which leads to a more productive conversation.
Lastly, I started what I call a new Pavlov home routine. When I’m done with work, I light a candle and write out three things I’m grateful for. Retrain your brain. Retrain your triggers. What’s your Pavlov’s bell going to be? Many of us come home hungry and stressed. Food then becomes linked to stress. This is not good. Link it with something else. Light a candle, count to 3, then blow it out. Use your kids to incorporate something fun. Use a toy with “super powers” to “beam” the bad feelings away. Taking a few extra minutes to shift gears has created a much happier home for me.
There are things that we can’t control. That’s called circumstances. We can’t control other people; we can’t control the hospital system; we can’t control our past. But the rest of everything we can control: our thoughts, feelings, and daily self-care rituals.
It reminds me of something my dad always said when I was a little girl. When crossing the street, you always look twice, oftentimes three. Why be so careful? It’s the pedestrian’s right of way after all. “Well..” he replied, “If a car hits you, nothing much happens to them, but your entire life will be destroyed, forever.”
Stop walking into traffic thinking everything will be ok. Take control of what you can.
Look, I get it. As health-care providers, we are an independent group. But just because you can do it alone, doesn’t mean you have to.
Choose one thing. Whether it be something I mentioned or something that came to your mind as you read this. Then, drop me a line at my personal email roozehra.khan.do@gmail.com. I will send you a reply to let you know I hear you and I’m in your corner.
Burnout happens.
But, so does joy, job satisfaction, and balance. Those things just take more effort.
Dr. Khan is Assistant Editor, Web and Multimedia, CHEST® journal.
Does this sound like your day?
You show up to work after a terrible night’s sleep. Your back is tense, and you do some kind of walking/stretching combo as you walk through the doors. Your focus fades during the mind-numbing routine of the morning shift sign out. As the day moves forward, you begin to feel resentful as you sign orders, see patients, and address your ICU team needs. You know that’s not right, that it’s not in line with who you want to be, but the irritation doesn’t go away.
Your lunchtime is filled with computer screens, notes, billing, and more billing. The previous feelings of irritation begin to boil into anger because more of your day is filled with bureaucratic demands and insurance reports rather than actually helping people. This isn’t what you signed up for. Years and years of training so you could be a paper pusher? The thought leads to rage ... or sometimes apathy on days you give in to the inevitable.
You finish your shift with admissions, procedures, code blues, and an overwhelming and exhausting night shift sign out. You feel like a hamster in a wheel. You’re going nowhere. What’s the point of all of this? You find yourself questioning why you went into medicine anyways ... yeah, that’s burnout.
I know what you’re thinking. You keep hearing about this, and it’s important to recognize, but then you hear the same old solutions: be more positive, find balance, do some yoga, take this resilience module, be mindful (what on earth does this mean anyways?), get some more sleep. Basically, it’s our problem. It’s our burden. If all of these were easy to understand and implement, don’t you think doctors and health-care providers would have done it already? I think you and I are a lot alike. These were my exact feelings. But stick with me on this one. I have a solution for you, albeit a little different. I’ll show you a more “positive” spin on the DIY.
I burned out early. After fellowship, I didn’t want to be a doctor anymore. I desperately sought to alter my career somehow. I looked into website development, something I had been good at in high school. I took a few refresher classes on my days off and started coding my own sites, but I had bills to pay. Big bills. Student loan bills. Luckily, my first job out of fellowship accepted many of my schedule demands, such as day shifts only, and after about a year, I recovered and remembered why I had loved medicine to begin with.
What is burnout?
Mind-body-soul exhaustion caused by excessive stress. Stress and burnout are closely related, but they’re more like distant cousins. Stress can be (and is) a normal part of our jobs. I bet you think you’re stressed, when you’re probably burned out. Critical care doctors have the highest rate of burnout among all physician subspecialties at >55%, and it is even higher in pediatric critical care. (Sessler C. https://www.mdedge.com/chestphysician/article/160951/society-news/turning-heat-icu-burnout). The main difference between stress and burnout is hope. With stress, you still feel like things can get better and you can get it all under control. Burnout feels hopeless.
What are the three core symptoms of burnout?
• Irritability and impatience with patients (depersonalization)
• Cynicism and difficulty concentrating (emotional exhaustion)
• What’s the point of all of this? Nothing I do matters or is appreciated (decreased self-efficacy)
We can talk about the symptoms of burnout all day, but what does that really look like? It looks like the day we described at the beginning. You know, the day that resonated with you and caused you to keep reading.
Why should we all be discussing this important topic?
Being burned out not only affects us on a soul level (achingly described above), but, more importantly, this can trickle down to our personal lives, family relationships, and how we care for our patients, with some studies showing that it affects our performance and, gulp, patient outcomes. That’s scary (Moss M et al. Crit Care Med. 2016;44[7]:1414).
Causes of burnout
There are many causes of burnout, and several studies have identified risk factors. A lack of control, conflicts with colleagues and leadership, and performing menial tasks can add to the irritation of a workday. This doesn’t even include the nature of our actual job as critical care doctors. We care for the sickest and are frequently involved in end-of-life care. Over time, the stress morphs into burnout. Female gender is also an independent risk factor for doctors (Pastores SM, et al. Crit Care Med. 2019;47[4]:550).
We’ve identified it. We’ve quantified it. But we’re not fixing it. In fact, there are only a few studies that have incorporated a needs assessment of doctors, paired with appropriate environmental intervention. A study done with primary care doctors in New York City clinics found that surveying a doctor’s “wish list” of interventions can help identify gaps in workflow, such as pairing one medical assistant with each attending (Linzer M, et al. J Gen Intern Med. 2015;30[8]:1105).
Without more data like this, we’re hamsters in a wheel. Luckily, organizations like CHEST have joined together with others to create the Critical Care Societies Collaborative and have an annual summit to discuss research strategies.
Solutions
Even millennials are sick of the mindful “chore” list. Yoga pants, yoga mats, crystals, chakras, meditation, and the list goes on and on. What millennials want are work-life integrations that are easy; workspaces that invite mindful behavior and daily rituals that excite and relax them. Co-working spaces like WeWork have designated self-care spaces.
Self-care is now essential, not an indulgence. I wasn’t sure how to create this space in my ICU, so I started small, with things I could carry with myself. The key is to find small rituals with big meanings. What could this look like for you? I began doing breathwork. Frankly, the idea came to me from my Apple® watch. It just started giving me these reminders one day, and I decided to take it seriously. I found that my mind and muscles eased after only 1 minute of breathing in and out slowly. This elevated my mood and was the refresher I needed in the afternoons. My body ached less after procedures.
I also got a little woo-woo (stay with me now) and began carrying around crystal stones. You don’t have to carry around crystals. Prayer books, religious symbols, your child’s toy car, anything can work if it has meaning for you, so when you see it or touch it during your day, you remember your big why. Why you’re serving people. Why you’re a doctor. I prefer the crystals over jewelry because it’s something unusual that I don’t expect to be sitting in my pocket. It’s always a nice gentle reminder of the love I have for my patients, my job, and humanity. When I put my hands in my pocket as I’m talking to yet another frustrated family member, my responses are more patient and calmer, which leads to a more productive conversation.
Lastly, I started what I call a new Pavlov home routine. When I’m done with work, I light a candle and write out three things I’m grateful for. Retrain your brain. Retrain your triggers. What’s your Pavlov’s bell going to be? Many of us come home hungry and stressed. Food then becomes linked to stress. This is not good. Link it with something else. Light a candle, count to 3, then blow it out. Use your kids to incorporate something fun. Use a toy with “super powers” to “beam” the bad feelings away. Taking a few extra minutes to shift gears has created a much happier home for me.
There are things that we can’t control. That’s called circumstances. We can’t control other people; we can’t control the hospital system; we can’t control our past. But the rest of everything we can control: our thoughts, feelings, and daily self-care rituals.
It reminds me of something my dad always said when I was a little girl. When crossing the street, you always look twice, oftentimes three. Why be so careful? It’s the pedestrian’s right of way after all. “Well..” he replied, “If a car hits you, nothing much happens to them, but your entire life will be destroyed, forever.”
Stop walking into traffic thinking everything will be ok. Take control of what you can.
Look, I get it. As health-care providers, we are an independent group. But just because you can do it alone, doesn’t mean you have to.
Choose one thing. Whether it be something I mentioned or something that came to your mind as you read this. Then, drop me a line at my personal email roozehra.khan.do@gmail.com. I will send you a reply to let you know I hear you and I’m in your corner.
Burnout happens.
But, so does joy, job satisfaction, and balance. Those things just take more effort.
Dr. Khan is Assistant Editor, Web and Multimedia, CHEST® journal.
Does this sound like your day?
You show up to work after a terrible night’s sleep. Your back is tense, and you do some kind of walking/stretching combo as you walk through the doors. Your focus fades during the mind-numbing routine of the morning shift sign out. As the day moves forward, you begin to feel resentful as you sign orders, see patients, and address your ICU team needs. You know that’s not right, that it’s not in line with who you want to be, but the irritation doesn’t go away.
Your lunchtime is filled with computer screens, notes, billing, and more billing. The previous feelings of irritation begin to boil into anger because more of your day is filled with bureaucratic demands and insurance reports rather than actually helping people. This isn’t what you signed up for. Years and years of training so you could be a paper pusher? The thought leads to rage ... or sometimes apathy on days you give in to the inevitable.
You finish your shift with admissions, procedures, code blues, and an overwhelming and exhausting night shift sign out. You feel like a hamster in a wheel. You’re going nowhere. What’s the point of all of this? You find yourself questioning why you went into medicine anyways ... yeah, that’s burnout.
I know what you’re thinking. You keep hearing about this, and it’s important to recognize, but then you hear the same old solutions: be more positive, find balance, do some yoga, take this resilience module, be mindful (what on earth does this mean anyways?), get some more sleep. Basically, it’s our problem. It’s our burden. If all of these were easy to understand and implement, don’t you think doctors and health-care providers would have done it already? I think you and I are a lot alike. These were my exact feelings. But stick with me on this one. I have a solution for you, albeit a little different. I’ll show you a more “positive” spin on the DIY.
I burned out early. After fellowship, I didn’t want to be a doctor anymore. I desperately sought to alter my career somehow. I looked into website development, something I had been good at in high school. I took a few refresher classes on my days off and started coding my own sites, but I had bills to pay. Big bills. Student loan bills. Luckily, my first job out of fellowship accepted many of my schedule demands, such as day shifts only, and after about a year, I recovered and remembered why I had loved medicine to begin with.
What is burnout?
Mind-body-soul exhaustion caused by excessive stress. Stress and burnout are closely related, but they’re more like distant cousins. Stress can be (and is) a normal part of our jobs. I bet you think you’re stressed, when you’re probably burned out. Critical care doctors have the highest rate of burnout among all physician subspecialties at >55%, and it is even higher in pediatric critical care. (Sessler C. https://www.mdedge.com/chestphysician/article/160951/society-news/turning-heat-icu-burnout). The main difference between stress and burnout is hope. With stress, you still feel like things can get better and you can get it all under control. Burnout feels hopeless.
What are the three core symptoms of burnout?
• Irritability and impatience with patients (depersonalization)
• Cynicism and difficulty concentrating (emotional exhaustion)
• What’s the point of all of this? Nothing I do matters or is appreciated (decreased self-efficacy)
We can talk about the symptoms of burnout all day, but what does that really look like? It looks like the day we described at the beginning. You know, the day that resonated with you and caused you to keep reading.
Why should we all be discussing this important topic?
Being burned out not only affects us on a soul level (achingly described above), but, more importantly, this can trickle down to our personal lives, family relationships, and how we care for our patients, with some studies showing that it affects our performance and, gulp, patient outcomes. That’s scary (Moss M et al. Crit Care Med. 2016;44[7]:1414).
Causes of burnout
There are many causes of burnout, and several studies have identified risk factors. A lack of control, conflicts with colleagues and leadership, and performing menial tasks can add to the irritation of a workday. This doesn’t even include the nature of our actual job as critical care doctors. We care for the sickest and are frequently involved in end-of-life care. Over time, the stress morphs into burnout. Female gender is also an independent risk factor for doctors (Pastores SM, et al. Crit Care Med. 2019;47[4]:550).
We’ve identified it. We’ve quantified it. But we’re not fixing it. In fact, there are only a few studies that have incorporated a needs assessment of doctors, paired with appropriate environmental intervention. A study done with primary care doctors in New York City clinics found that surveying a doctor’s “wish list” of interventions can help identify gaps in workflow, such as pairing one medical assistant with each attending (Linzer M, et al. J Gen Intern Med. 2015;30[8]:1105).
Without more data like this, we’re hamsters in a wheel. Luckily, organizations like CHEST have joined together with others to create the Critical Care Societies Collaborative and have an annual summit to discuss research strategies.
Solutions
Even millennials are sick of the mindful “chore” list. Yoga pants, yoga mats, crystals, chakras, meditation, and the list goes on and on. What millennials want are work-life integrations that are easy; workspaces that invite mindful behavior and daily rituals that excite and relax them. Co-working spaces like WeWork have designated self-care spaces.
Self-care is now essential, not an indulgence. I wasn’t sure how to create this space in my ICU, so I started small, with things I could carry with myself. The key is to find small rituals with big meanings. What could this look like for you? I began doing breathwork. Frankly, the idea came to me from my Apple® watch. It just started giving me these reminders one day, and I decided to take it seriously. I found that my mind and muscles eased after only 1 minute of breathing in and out slowly. This elevated my mood and was the refresher I needed in the afternoons. My body ached less after procedures.
I also got a little woo-woo (stay with me now) and began carrying around crystal stones. You don’t have to carry around crystals. Prayer books, religious symbols, your child’s toy car, anything can work if it has meaning for you, so when you see it or touch it during your day, you remember your big why. Why you’re serving people. Why you’re a doctor. I prefer the crystals over jewelry because it’s something unusual that I don’t expect to be sitting in my pocket. It’s always a nice gentle reminder of the love I have for my patients, my job, and humanity. When I put my hands in my pocket as I’m talking to yet another frustrated family member, my responses are more patient and calmer, which leads to a more productive conversation.
Lastly, I started what I call a new Pavlov home routine. When I’m done with work, I light a candle and write out three things I’m grateful for. Retrain your brain. Retrain your triggers. What’s your Pavlov’s bell going to be? Many of us come home hungry and stressed. Food then becomes linked to stress. This is not good. Link it with something else. Light a candle, count to 3, then blow it out. Use your kids to incorporate something fun. Use a toy with “super powers” to “beam” the bad feelings away. Taking a few extra minutes to shift gears has created a much happier home for me.
There are things that we can’t control. That’s called circumstances. We can’t control other people; we can’t control the hospital system; we can’t control our past. But the rest of everything we can control: our thoughts, feelings, and daily self-care rituals.
It reminds me of something my dad always said when I was a little girl. When crossing the street, you always look twice, oftentimes three. Why be so careful? It’s the pedestrian’s right of way after all. “Well..” he replied, “If a car hits you, nothing much happens to them, but your entire life will be destroyed, forever.”
Stop walking into traffic thinking everything will be ok. Take control of what you can.
Look, I get it. As health-care providers, we are an independent group. But just because you can do it alone, doesn’t mean you have to.
Choose one thing. Whether it be something I mentioned or something that came to your mind as you read this. Then, drop me a line at my personal email roozehra.khan.do@gmail.com. I will send you a reply to let you know I hear you and I’m in your corner.
Burnout happens.
But, so does joy, job satisfaction, and balance. Those things just take more effort.
Dr. Khan is Assistant Editor, Web and Multimedia, CHEST® journal.
Risks of removing the default: Lung protective ventilation IS for everyone
Since the landmark ARMA trial, use of low tidal volume ventilation (LTVV) at 6 mL/kg predicted body weight (PBW) has become our gold standard for ventilator management in acute respiratory distress syndrome (ARDS) (Brower RG, et al. N Engl J Med. 2000;342[18]:1301). While other studies have suggested that patients without ARDS may also benefit from lower volumes, the recently published Protective Ventilation in Patients Without ARDS (PReVENT) trial found no benefit to using LTVV in non-ARDS patients (Simonis FD, et al. JAMA. 2018;320[18]:1872). Does this mean we let physicians set volumes at will? Is tidal volume (VT) even clinically relevant anymore in the non-ARDS population?
Prior to the PReVENT trial, our practice of LTVV for patients without ARDS was informed primarily by observational data. In 2012, a meta-analysis comparing LTVV with “conventional” VT (10-12 mL/kg IBW) in non-ARDS patients found that those given LTVV had a lower incidence of acute lung injury and lower overall mortality (Neto AS, et al. JAMA. 2012 308[16]:1651). While these were promising findings, there was limited follow-up poststudy onset, and the majority of included studies were based on a surgical population. Additionally, the use of VT > 10 mL/kg PBW has become uncommon in routine clinical practice. How comparable are those previous studies to today’s clinical milieu? When comparing outcomes for ICU patients who were ventilated with low (≤7mL/kg PBW), intermediate (>7, but <10 mL/kg PBW), and high (≥10 mL/kg PBW) VT, a second meta-analysis found a 28% risk reduction in the development of ARDS or pneumonia with low vs high, but the similar difference was not seen when comparing low vs intermediate groups (Neto AS, et al. Crit Care Med. 2015;43[10]:2155). This research suggested that negative outcomes were driven by the excessive VT.

Slated to be the definitive study on the matter, the PReVENT trial used a multicenter randomized control trial design comparing target VT of 4 mL/kg with 10 mL/kg PBW, with setting titration primarily based on plateau pressure targets. The headline out of this trial may have been that it was “negative,” in that there was no difference between the groups in the primary outcome of ventilator-free days and survival by day 28. However, there are some important limitations to consider before discounting LTVV for everyone. First, half of the trial patients were ventilated with pressure-control ventilation, the actual VT settings were 7.3 (5.9 – 9.1) for the low group vs 9.1 (7.7 – 10.5) mL/kg PBW for the intermediate group by day 3, statistically significant differences, but perhaps not as striking clinically. Moreover, a secondary analysis of ARDSnet data (Amato MB, et al, N Engl J Med. 2015;372[8]:747) also suggests that driving pressure, more so than VT, may determine outcomes, which, for most patients in the PReVENT trial, remained in the “safe” range of < 15 cm H2O. Finally, almost two-thirds of patients eligible for PReVENT were not enrolled, and the included cohort had PaO2/FiO2 ratios greater than 200 for the 3 days of the study, limiting generalizability, especially for patients with acute hypoxemic respiratory failure.
When approaching the patient who we have determined to not have ARDS (either by clinical diagnosis or suspicion plus a low PaO2/FiO2 ratio as defined by PReVENT’s protocol), it is important to also consider our accuracy in recognizing ARDS before settling for the use of unregulated VT. ARDS is often underrecognized, and this delay in diagnosis results in delayed LTVV initiation. Results from the LUNG SAFE study, an international multicenter prospective observational study of over 2,300 ICU patients with ARDS, showed that only 34% of patients were recognized by the clinician to have ARDS at the time they met the Berlin criteria (Bellani G, et al. JAMA. 2016;315[8]:788). As ARDS is defined by clinical criteria, it is biologically plausible to think that the pathologic process commences before these criteria are recognized by the clinician.

To investigate the importance of timing of LTVV in ARDS, Needham and colleagues performed a prospective cohort study in patients with ARDS, examining the effect of VT received over time on the outcome of ICU mortality (Needham DM, et al. Am J Respir Crit Care Med. 2015;191[2]:177). They found that every 1 mL/kg increase in VT setting was associated with a 23% increase in mortality and, indeed, increases in subsequent VT compared with baseline setting were associated with increasing mortality. One may, therefore, be concerned that if we miss the ARDS diagnosis, the default to higher VT at the time of intubation may harm our patients. With or without clinician recognition of ARDS, LUNG SAFE revealed that the average VT for the patients with confirmed ARDS was 7.6 (95% CI 7.5-7.7) mL/kg PBW. While this mean value is well within the range of lung protective ventilation (less than 8 mL/kg PBW), over one-third of patients were exposed to larger VT. A recently published study by Sjoding and colleagues showed that VT of >8 mL/kg PBW was used in 40% of the cohort, and continued exposure to 24 total hours of these high VT was associated with increased risk of mortality (OR 1.82 (95% CI, 1.20–2.78) (Sjoding MW, et al. Crit Care Med. 2019;47[1]:56). All three studies support early administration of lung protective ventilation, considering the high mortality associated with ARDS.
Before consolidating what we know about empiric use of LTVV, we also must highlight the important concerns about LTVV that were investigated in the PReVENT trial. Over-sedation to maintain low VT, increased delirium, ventilator asynchrony, and possibility of effort-induced lung injury are some of the potential risks associated with LTVV. While there were no differences in the use of sedatives or neuromuscular blocking agents between groups in the PReVENT trial, more delirium was seen in the LTVV group with a P = .06, which may be a signal deserving further exploration.
Therefore, now understanding both the upside and downside of LTVV, what’s our best approach? While we lack prospective clinical trial data showing benefit of LTVV in patients without ARDS, we do not have conclusive evidence to show its harm. Remembering that even intensivists can fail to recognize ARDS at its onset, default utilization of LTVV, or at least lung protective ventilation of <8 mL/kg PBW, may be the safest approach for all patients. To be clear, this approach would still allow for active physician decision-making to personalize the settings to the individual patient’s needs, including the use of higher VT if needed for patient comfort, effort, and sedation needs. Changing the default settings and implementing friendly reminders about how to manage the ventilator has already been shown to be helpful for the surgical population (O’Reilly-Shah VN, et al. BMJ Qual Saf. 2018;27[12]:1008).
We must also consider the process of health-care delivery and the implementation of best practices, after considering the facilitators and barriers to adoption of said practices. Many patients decompensate and require intubation prior to ICU arrival, with prolonged boarding in the ED or medical wards being a common occurrence for many hospitals. As such, we need to consider a ventilation strategy that allows for best practice implementation at a hospital-wide level, appealing to an interprofessional approach to ventilator management, employing physicians outside of critical care medicine, respiratory therapists, and nursing. The PReVENT trial had a nicely constructed protocol with clear instructions on ventilator adjustments with frequent plateau pressure measurements and patient assessments. In the real world setting, especially in a non-ICU setting, ventilator management is not as straightforward. Considering that plateau pressures were only checked in approximately 40% of the patients in LUNG SAFE cohort, active management and attention to driving pressure may be a stretch in many settings.
Until we get 100% sensitive in timely recognition (instantaneous, really) of ARDS pathology augmented by automated diagnostic tools embedded in the medical record and/or incorporate advanced technology in the ventilator management to avoid human error, employing simple defaults to guarantee a protective setting in case of later diagnosis of ARDS seems logical. We can even go further to separate the defaults into LTVV for hypoxemic respiratory failure and lung protective ventilation for everything else, with future development of more algorithms, protocols, and clinical decision support tools for ventilator management. For the time being, a simpler intervention of setting a safer default is a great universal start.
Dr. Mathews and Dr. Howell are with the Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine; Dr. Mathews is also with the Department of Emergency Medicine; Icahn School of Medicine at Mount Sinai, New York, NY.
Since the landmark ARMA trial, use of low tidal volume ventilation (LTVV) at 6 mL/kg predicted body weight (PBW) has become our gold standard for ventilator management in acute respiratory distress syndrome (ARDS) (Brower RG, et al. N Engl J Med. 2000;342[18]:1301). While other studies have suggested that patients without ARDS may also benefit from lower volumes, the recently published Protective Ventilation in Patients Without ARDS (PReVENT) trial found no benefit to using LTVV in non-ARDS patients (Simonis FD, et al. JAMA. 2018;320[18]:1872). Does this mean we let physicians set volumes at will? Is tidal volume (VT) even clinically relevant anymore in the non-ARDS population?
Prior to the PReVENT trial, our practice of LTVV for patients without ARDS was informed primarily by observational data. In 2012, a meta-analysis comparing LTVV with “conventional” VT (10-12 mL/kg IBW) in non-ARDS patients found that those given LTVV had a lower incidence of acute lung injury and lower overall mortality (Neto AS, et al. JAMA. 2012 308[16]:1651). While these were promising findings, there was limited follow-up poststudy onset, and the majority of included studies were based on a surgical population. Additionally, the use of VT > 10 mL/kg PBW has become uncommon in routine clinical practice. How comparable are those previous studies to today’s clinical milieu? When comparing outcomes for ICU patients who were ventilated with low (≤7mL/kg PBW), intermediate (>7, but <10 mL/kg PBW), and high (≥10 mL/kg PBW) VT, a second meta-analysis found a 28% risk reduction in the development of ARDS or pneumonia with low vs high, but the similar difference was not seen when comparing low vs intermediate groups (Neto AS, et al. Crit Care Med. 2015;43[10]:2155). This research suggested that negative outcomes were driven by the excessive VT.
Slated to be the definitive study on the matter, the PReVENT trial used a multicenter randomized control trial design comparing target VT of 4 mL/kg with 10 mL/kg PBW, with setting titration primarily based on plateau pressure targets. The headline out of this trial may have been that it was “negative,” in that there was no difference between the groups in the primary outcome of ventilator-free days and survival by day 28. However, there are some important limitations to consider before discounting LTVV for everyone. First, half of the trial patients were ventilated with pressure-control ventilation, the actual VT settings were 7.3 (5.9 – 9.1) for the low group vs 9.1 (7.7 – 10.5) mL/kg PBW for the intermediate group by day 3, statistically significant differences, but perhaps not as striking clinically. Moreover, a secondary analysis of ARDSnet data (Amato MB, et al, N Engl J Med. 2015;372[8]:747) also suggests that driving pressure, more so than VT, may determine outcomes, which, for most patients in the PReVENT trial, remained in the “safe” range of < 15 cm H2O. Finally, almost two-thirds of patients eligible for PReVENT were not enrolled, and the included cohort had PaO2/FiO2 ratios greater than 200 for the 3 days of the study, limiting generalizability, especially for patients with acute hypoxemic respiratory failure.
When approaching the patient who we have determined to not have ARDS (either by clinical diagnosis or suspicion plus a low PaO2/FiO2 ratio as defined by PReVENT’s protocol), it is important to also consider our accuracy in recognizing ARDS before settling for the use of unregulated VT. ARDS is often underrecognized, and this delay in diagnosis results in delayed LTVV initiation. Results from the LUNG SAFE study, an international multicenter prospective observational study of over 2,300 ICU patients with ARDS, showed that only 34% of patients were recognized by the clinician to have ARDS at the time they met the Berlin criteria (Bellani G, et al. JAMA. 2016;315[8]:788). As ARDS is defined by clinical criteria, it is biologically plausible to think that the pathologic process commences before these criteria are recognized by the clinician.
To investigate the importance of timing of LTVV in ARDS, Needham and colleagues performed a prospective cohort study in patients with ARDS, examining the effect of VT received over time on the outcome of ICU mortality (Needham DM, et al. Am J Respir Crit Care Med. 2015;191[2]:177). They found that every 1 mL/kg increase in VT setting was associated with a 23% increase in mortality and, indeed, increases in subsequent VT compared with baseline setting were associated with increasing mortality. One may, therefore, be concerned that if we miss the ARDS diagnosis, the default to higher VT at the time of intubation may harm our patients. With or without clinician recognition of ARDS, LUNG SAFE revealed that the average VT for the patients with confirmed ARDS was 7.6 (95% CI 7.5-7.7) mL/kg PBW. While this mean value is well within the range of lung protective ventilation (less than 8 mL/kg PBW), over one-third of patients were exposed to larger VT. A recently published study by Sjoding and colleagues showed that VT of >8 mL/kg PBW was used in 40% of the cohort, and continued exposure to 24 total hours of these high VT was associated with increased risk of mortality (OR 1.82 (95% CI, 1.20–2.78) (Sjoding MW, et al. Crit Care Med. 2019;47[1]:56). All three studies support early administration of lung protective ventilation, considering the high mortality associated with ARDS.
Before consolidating what we know about empiric use of LTVV, we also must highlight the important concerns about LTVV that were investigated in the PReVENT trial. Over-sedation to maintain low VT, increased delirium, ventilator asynchrony, and possibility of effort-induced lung injury are some of the potential risks associated with LTVV. While there were no differences in the use of sedatives or neuromuscular blocking agents between groups in the PReVENT trial, more delirium was seen in the LTVV group with a P = .06, which may be a signal deserving further exploration.
Therefore, now understanding both the upside and downside of LTVV, what’s our best approach? While we lack prospective clinical trial data showing benefit of LTVV in patients without ARDS, we do not have conclusive evidence to show its harm. Remembering that even intensivists can fail to recognize ARDS at its onset, default utilization of LTVV, or at least lung protective ventilation of <8 mL/kg PBW, may be the safest approach for all patients. To be clear, this approach would still allow for active physician decision-making to personalize the settings to the individual patient’s needs, including the use of higher VT if needed for patient comfort, effort, and sedation needs. Changing the default settings and implementing friendly reminders about how to manage the ventilator has already been shown to be helpful for the surgical population (O’Reilly-Shah VN, et al. BMJ Qual Saf. 2018;27[12]:1008).
We must also consider the process of health-care delivery and the implementation of best practices, after considering the facilitators and barriers to adoption of said practices. Many patients decompensate and require intubation prior to ICU arrival, with prolonged boarding in the ED or medical wards being a common occurrence for many hospitals. As such, we need to consider a ventilation strategy that allows for best practice implementation at a hospital-wide level, appealing to an interprofessional approach to ventilator management, employing physicians outside of critical care medicine, respiratory therapists, and nursing. The PReVENT trial had a nicely constructed protocol with clear instructions on ventilator adjustments with frequent plateau pressure measurements and patient assessments. In the real world setting, especially in a non-ICU setting, ventilator management is not as straightforward. Considering that plateau pressures were only checked in approximately 40% of the patients in LUNG SAFE cohort, active management and attention to driving pressure may be a stretch in many settings.
Until we get 100% sensitive in timely recognition (instantaneous, really) of ARDS pathology augmented by automated diagnostic tools embedded in the medical record and/or incorporate advanced technology in the ventilator management to avoid human error, employing simple defaults to guarantee a protective setting in case of later diagnosis of ARDS seems logical. We can even go further to separate the defaults into LTVV for hypoxemic respiratory failure and lung protective ventilation for everything else, with future development of more algorithms, protocols, and clinical decision support tools for ventilator management. For the time being, a simpler intervention of setting a safer default is a great universal start.
Dr. Mathews and Dr. Howell are with the Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine; Dr. Mathews is also with the Department of Emergency Medicine; Icahn School of Medicine at Mount Sinai, New York, NY.
Since the landmark ARMA trial, use of low tidal volume ventilation (LTVV) at 6 mL/kg predicted body weight (PBW) has become our gold standard for ventilator management in acute respiratory distress syndrome (ARDS) (Brower RG, et al. N Engl J Med. 2000;342[18]:1301). While other studies have suggested that patients without ARDS may also benefit from lower volumes, the recently published Protective Ventilation in Patients Without ARDS (PReVENT) trial found no benefit to using LTVV in non-ARDS patients (Simonis FD, et al. JAMA. 2018;320[18]:1872). Does this mean we let physicians set volumes at will? Is tidal volume (VT) even clinically relevant anymore in the non-ARDS population?
Prior to the PReVENT trial, our practice of LTVV for patients without ARDS was informed primarily by observational data. In 2012, a meta-analysis comparing LTVV with “conventional” VT (10-12 mL/kg IBW) in non-ARDS patients found that those given LTVV had a lower incidence of acute lung injury and lower overall mortality (Neto AS, et al. JAMA. 2012 308[16]:1651). While these were promising findings, there was limited follow-up poststudy onset, and the majority of included studies were based on a surgical population. Additionally, the use of VT > 10 mL/kg PBW has become uncommon in routine clinical practice. How comparable are those previous studies to today’s clinical milieu? When comparing outcomes for ICU patients who were ventilated with low (≤7mL/kg PBW), intermediate (>7, but <10 mL/kg PBW), and high (≥10 mL/kg PBW) VT, a second meta-analysis found a 28% risk reduction in the development of ARDS or pneumonia with low vs high, but the similar difference was not seen when comparing low vs intermediate groups (Neto AS, et al. Crit Care Med. 2015;43[10]:2155). This research suggested that negative outcomes were driven by the excessive VT.
Slated to be the definitive study on the matter, the PReVENT trial used a multicenter randomized control trial design comparing target VT of 4 mL/kg with 10 mL/kg PBW, with setting titration primarily based on plateau pressure targets. The headline out of this trial may have been that it was “negative,” in that there was no difference between the groups in the primary outcome of ventilator-free days and survival by day 28. However, there are some important limitations to consider before discounting LTVV for everyone. First, half of the trial patients were ventilated with pressure-control ventilation, the actual VT settings were 7.3 (5.9 – 9.1) for the low group vs 9.1 (7.7 – 10.5) mL/kg PBW for the intermediate group by day 3, statistically significant differences, but perhaps not as striking clinically. Moreover, a secondary analysis of ARDSnet data (Amato MB, et al, N Engl J Med. 2015;372[8]:747) also suggests that driving pressure, more so than VT, may determine outcomes, which, for most patients in the PReVENT trial, remained in the “safe” range of < 15 cm H2O. Finally, almost two-thirds of patients eligible for PReVENT were not enrolled, and the included cohort had PaO2/FiO2 ratios greater than 200 for the 3 days of the study, limiting generalizability, especially for patients with acute hypoxemic respiratory failure.
When approaching the patient who we have determined to not have ARDS (either by clinical diagnosis or suspicion plus a low PaO2/FiO2 ratio as defined by PReVENT’s protocol), it is important to also consider our accuracy in recognizing ARDS before settling for the use of unregulated VT. ARDS is often underrecognized, and this delay in diagnosis results in delayed LTVV initiation. Results from the LUNG SAFE study, an international multicenter prospective observational study of over 2,300 ICU patients with ARDS, showed that only 34% of patients were recognized by the clinician to have ARDS at the time they met the Berlin criteria (Bellani G, et al. JAMA. 2016;315[8]:788). As ARDS is defined by clinical criteria, it is biologically plausible to think that the pathologic process commences before these criteria are recognized by the clinician.
To investigate the importance of timing of LTVV in ARDS, Needham and colleagues performed a prospective cohort study in patients with ARDS, examining the effect of VT received over time on the outcome of ICU mortality (Needham DM, et al. Am J Respir Crit Care Med. 2015;191[2]:177). They found that every 1 mL/kg increase in VT setting was associated with a 23% increase in mortality and, indeed, increases in subsequent VT compared with baseline setting were associated with increasing mortality. One may, therefore, be concerned that if we miss the ARDS diagnosis, the default to higher VT at the time of intubation may harm our patients. With or without clinician recognition of ARDS, LUNG SAFE revealed that the average VT for the patients with confirmed ARDS was 7.6 (95% CI 7.5-7.7) mL/kg PBW. While this mean value is well within the range of lung protective ventilation (less than 8 mL/kg PBW), over one-third of patients were exposed to larger VT. A recently published study by Sjoding and colleagues showed that VT of >8 mL/kg PBW was used in 40% of the cohort, and continued exposure to 24 total hours of these high VT was associated with increased risk of mortality (OR 1.82 (95% CI, 1.20–2.78) (Sjoding MW, et al. Crit Care Med. 2019;47[1]:56). All three studies support early administration of lung protective ventilation, considering the high mortality associated with ARDS.
Before consolidating what we know about empiric use of LTVV, we also must highlight the important concerns about LTVV that were investigated in the PReVENT trial. Over-sedation to maintain low VT, increased delirium, ventilator asynchrony, and possibility of effort-induced lung injury are some of the potential risks associated with LTVV. While there were no differences in the use of sedatives or neuromuscular blocking agents between groups in the PReVENT trial, more delirium was seen in the LTVV group with a P = .06, which may be a signal deserving further exploration.
Therefore, now understanding both the upside and downside of LTVV, what’s our best approach? While we lack prospective clinical trial data showing benefit of LTVV in patients without ARDS, we do not have conclusive evidence to show its harm. Remembering that even intensivists can fail to recognize ARDS at its onset, default utilization of LTVV, or at least lung protective ventilation of <8 mL/kg PBW, may be the safest approach for all patients. To be clear, this approach would still allow for active physician decision-making to personalize the settings to the individual patient’s needs, including the use of higher VT if needed for patient comfort, effort, and sedation needs. Changing the default settings and implementing friendly reminders about how to manage the ventilator has already been shown to be helpful for the surgical population (O’Reilly-Shah VN, et al. BMJ Qual Saf. 2018;27[12]:1008).
We must also consider the process of health-care delivery and the implementation of best practices, after considering the facilitators and barriers to adoption of said practices. Many patients decompensate and require intubation prior to ICU arrival, with prolonged boarding in the ED or medical wards being a common occurrence for many hospitals. As such, we need to consider a ventilation strategy that allows for best practice implementation at a hospital-wide level, appealing to an interprofessional approach to ventilator management, employing physicians outside of critical care medicine, respiratory therapists, and nursing. The PReVENT trial had a nicely constructed protocol with clear instructions on ventilator adjustments with frequent plateau pressure measurements and patient assessments. In the real world setting, especially in a non-ICU setting, ventilator management is not as straightforward. Considering that plateau pressures were only checked in approximately 40% of the patients in LUNG SAFE cohort, active management and attention to driving pressure may be a stretch in many settings.
Until we get 100% sensitive in timely recognition (instantaneous, really) of ARDS pathology augmented by automated diagnostic tools embedded in the medical record and/or incorporate advanced technology in the ventilator management to avoid human error, employing simple defaults to guarantee a protective setting in case of later diagnosis of ARDS seems logical. We can even go further to separate the defaults into LTVV for hypoxemic respiratory failure and lung protective ventilation for everything else, with future development of more algorithms, protocols, and clinical decision support tools for ventilator management. For the time being, a simpler intervention of setting a safer default is a great universal start.
Dr. Mathews and Dr. Howell are with the Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine; Dr. Mathews is also with the Department of Emergency Medicine; Icahn School of Medicine at Mount Sinai, New York, NY.
Renal replacement therapy in the ICU: Vexed questions and team dynamics
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
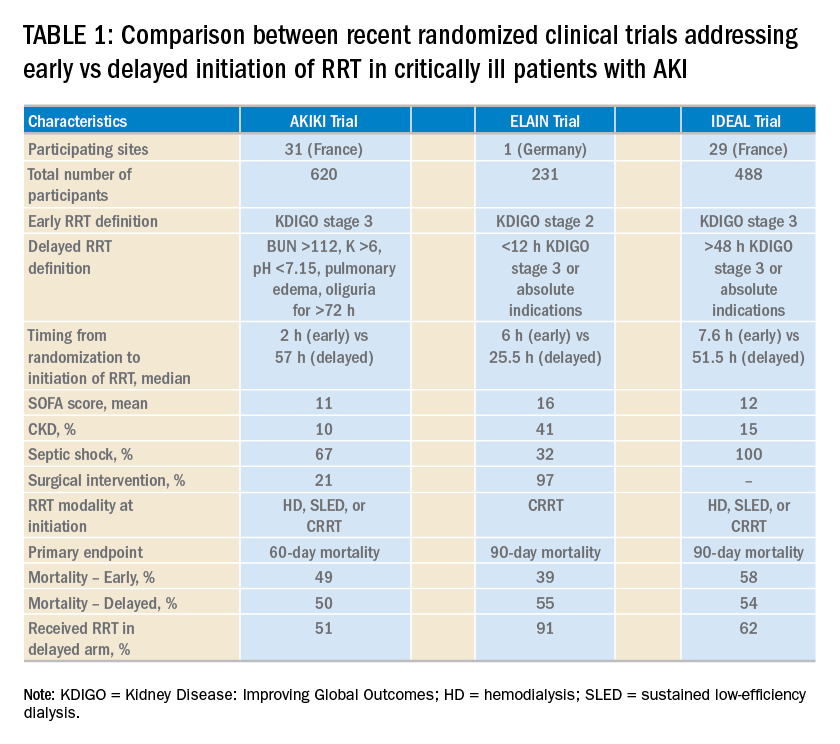
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?

The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?

The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?
The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?
The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
More than 5 million patients are admitted to ICUs each year in the United States, and approximately 2% to 10% of these patients develop acute kidney injury requiring renal replacement therapy (AKI-RRT). AKI-RRT carries high morbidity and mortality (Hoste EA, et al. Intensive Care Med. 2015;41:1411) and is associated with renal and systemic complications, such as cardiovascular disease. RRT, frequently provided by nephrologists and/or intensivists, is a supportive therapy that can be life-saving when provided to the right patient at the right time. However, several questions related to the provision of RRT still remain, including the optimal timing of RRT initiation, the development of quality metrics for optimal RRT deliverables and monitoring, and the optimal strategy of RRT de-escalation and risk-stratification of renal recovery. Overall, there is paucity of randomized trials and standardized risk-stratification tools that can guide RRT in the ICU.
Current vexed questions of RRT deliverables in the ICU
There is ongoing research aiming to answer critical questions that can potentially improve current standards of RRT.
What is the optimal time of RRT initiation for critically ill patients with AKI?
Over the last 2 years, three randomized clinical trials have attempted to address this important question involving heterogeneous ICU populations and distinct research hypotheses and study designs. Two of these studies, AKIKI (Gaudry S, et al. N Engl J Med. 2016;375:122) and IDEAL-ICU (Barbar SD, et al. N Engl J Med. 2018;379:1431) yielded no significant difference in the primary outcome of 60-day and 90-day all-cause mortality between the early vs delayed RRT initiation strategies, respectively (Table 1). Further, AKIKI showed no difference in RRT dependence at 60 days and higher catheter-related infections and hypophosphatemia in the early initiation arm. It is important to note that IDEAL-ICU was stopped early for futility after the second planned interim analysis with only 56% of patients enrolled (
How can RRT deliverables in the ICU be effectively and systematically monitored?
The provision of RRT to ICU patients with AKI requires an iterative adjustment of the RRT prescription and goals of therapy to accommodate changes in the clinical status with emphasis in hemodynamics, multiorgan failure, and fluid overload (Neyra JA. Clin Nephrol. 2018;90:1). The utilization of static and functional tests or point-of-care ultrasonography to assess hemodynamic variables can be useful. Furthermore, the implementation of customized and automated flowsheets in the electronic health record can facilitate remote monitoring. It is, therefore, essential that the multidisciplinary ICU team develops a process to monitor and ensure RRT deliverables. In this context, the standardization and monitoring of quality metrics (dose, modality, anticoagulation, filter life, downtime, etc) and the development of effective quality management systems are critically important. However, big multicenter data are direly needed to provide insight in this arena.
How can renal recovery be assessed and RRT effectively de-escalated?
The continuous examination of renal recovery in ICU patients with AKI-RRT is mostly based on urine output trend and, if feasible, interdialytic solute control. Sometimes, the transition from continuous RRT to intermittent modalities is necessary in the context of multiorgan recovery and de-escalation of care. However, clinical risk-prediction tools that identify patients who can potentially recover or already exhibit early signs of renal function recovery are needed. Current advances in clinical informatics can help to incorporate time-varying clinical parameters that may be informative for risk-prediction models. In addition, incorporating novel biomarkers of AKI repair and functional tests (eg, furosemide stress test, functional MRI) into these models may further inform these tools and aid the development of clinical decision support systems that enhance interventions to promote AKI recovery (Neyra JA, et al. Nephron. 2018;140: 99).
Is post-AKI outpatient care beneficial for ICU survivors who suffered from AKI-RRT?
Specialized AKI survivor clinics have been implemented in some centers. In general, this outpatient follow-up model includes survivors who suffered from AKI stage 2 or 3, some of them requiring RRT, and tailors individualized interventions for post-AKI complications (preventing recurrent AKI, attenuating incident or progressive CKD). However, the value of this outpatient model needs to be further evaluated with emphasis on clinical outcomes (eg, recurrent AKI, CKD, readmissions, or death) and elements that impact quality of life. This is an area of evolving research and a great opportunity for the nephrology and critical care communities to integrate and enhance post-ICU outpatient care and research collaboration.
Interdisciplinary communication among acute care team members
Two essential elements to provide effective RRT to ICU patients with AKI are: (1) the dynamics of the ICU team (intensivists, nephrologists, pharmacists, nurses, nutritionists, physical therapists, etc) to enhance the delivery of personalized therapy (RRT candidacy, timing of initiation, goals for solute control and fluid removal/regulation, renal recovery evaluation, RRT de-escalation, etc.) and (2) the frequent assessment and adjustment of RRT goals according to the clinical status of the patient. Therefore, effective RRT provision in the ICU requires the development of optimal channels of communication among all members of the acute care team and the systematic monitoring of the clinical status of the patient and RRT-specific goals and deliverables.
Perspective from a nurse and quality improvement officer for the provision of RRT in the ICU
The provision of continuous RRT (CRRT) to critically ill patients requires close communication between the bedside nurse and the rest of the ICU team. The physician typically prescribes CRRT and determines the specific goals of therapy. The pharmacist works closely with the nephrologist/intensivist and bedside nurse, especially in regards to customized CRRT solutions (when indicated) and medication dosing. Because CRRT can alter drug pharmacokinetics, the pharmacist closely and constantly monitors the patient’s clinical status, CRRT prescription, and all active medications. CRRT can also affect the nutritional and metabolic status of critically ill patients; therefore, the input of the nutritionist is necessary. The syndrome of ICU-acquired weakness is commonly encountered in ICU patients and is related to physical immobility. While ICU patients with AKI are already at risk for decreased mobility, the continuous connection to an immobile extracorporeal machine for the provision of CRRT may further contribute to immobilization and can also preclude the provision of optimal physical therapy. Therefore, the bedside nurse should assist the physical therapist for the timely and effective delivery of physical therapy according to the clinical status of the patient.
The clinical scenarios discussed above provide a small glimpse into the importance of developing an interdisciplinary ICU team caring for critically ill patients receiving CRRT. In the context of how integral the specific role of each team member is, it becomes clear that the bedside nurse’s role is not only to deliver hands-on patient care but also the orchestration of collaborative communication among all health-care providers for the effective provision of CRRT to critically ill patients in the ICU.
Dr. Neyra and Ms. Hauschild are with the Department of Internal Medicine; Division of Nephrology; Bone and Mineral Metabolism; University of Kentucky; Lexington, Kentucky.
The 1-hour sepsis bundle is serious—serious like a heart attack
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.

In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.
In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.
In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
ECMO for ARDS in the modern era
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.
Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.
Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.
Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
Balanced crystalloids vs saline for critically ill patients
If you work in an ICU, chances are good that you frequently order IV fluids (IVF). Between resuscitation, maintenance, and medication carriers, nearly all ICU patients receive IVF. Historically, much of this IVF has been 0.9% sodium chloride (“saline” or “normal saline”). Providers in the United States alone administer more than 200 million liters of saline each year (Myburgh JA, et al. N Engl J Med. 2013;369[13]:1243). New evidence, however, suggests that treating your ICU patients with so-called “balanced crystalloids,” rather than saline, may improve patient outcomes.

For over a century, clinicians ordering IV isotonic crystalloids have had two basic options: saline or balanced crystalloids (BC). Saline contains water and 154 mmol/L of sodium chloride (around 50% more chloride than human extracellular fluid). In contrast, BCs, like lactated Ringer’s (LR), Hartman’s solution, and others, contain an amount of chloride resembling human plasma (Table 1). BC substitute an organic anion such as bicarbonate, lactate, acetate, or gluconate, in place of chloride – resulting in lower chloride level and a more neutral pH.
Over the last 2 decades, evidence has slowly accumulated that the different compositions of saline and BC might translate into differences in patient physiology and outcomes. Research in the operating room and ICU found that saline administration caused hyperchloremia and metabolic acidosis. Studies of healthy volunteers found that saline decreased blood flow to the kidney (Chowdhury AH, et al. Ann Surg. 2012;256[1]:18). Animal sepsis models suggested that saline might cause inflammation, low blood pressure, and kidney injury (Zhou F, et al. Crit Care Med. 2014;42[4]:e270). Large observational studies among ICU patients found saline to be associated with increased risk of kidney injury, dialysis, or death (Raghunathan K, et al. Crit Care Med. 2014 Jul;42[7]:1585). These preliminary studies set the stage for a large randomized clinical trial comparing clinical outcomes between BC and saline among acutely ill adults.
Between June 2015 and April 2017, our research group conducted the Isotonic Solutions and Major Adverse Renal Events Trial (SMART) (Semler MW, et al. N Engl J Med. 2018;378[9]:819). SMART was a pragmatic trial in which 15,802 adults in five ICUs were assigned to receive either saline (0.9% sodium chloride) or BC (LR or another branded BC [PlasmaLyte A]). The goal was to determine whether using BC rather than saline would decrease the rates of death, new dialysis, or renal dysfunction lasting through hospital discharge. Patients in the BC group received primarily BC (44% LR and 56% another branded BC [PlasmaLyte A]), whereas patients in the saline group received primarily saline. The rate of death, new dialysis, or renal dysfunction lasting through hospital discharge was lower in the BC group (14.3%) than the saline group (15.4%) (OR: 0.90; 95% CI, 0.82-0.99; P=0.04). The difference between groups was primarily in death and new dialysis, not changes in creatinine. For every 100 patients admitted to an ICU, using BC rather than saline would spare one patient from experiencing death, dialysis, or renal dysfunction lasting to hospital discharge (number needed to treat). The benefits of BC appeared to be greater among patients who received larger volumes of IVF and patients with sepsis. In fact, among patients with sepsis, mortality was significantly lower with BC (25.2%) than with saline (29.4%) (P=.02).
Another trial was conducted in parallel. Saline against LR or another branded BC (PlasmaLyte) in the ED (SALT-ED) compared BC with saline among 13,347 non-critically ill adults treated with IVF in the ED (Self WH, et al. N Engl J Med. 2018;378[9]:829). Like the SMART trial, the SALT-ED trial found a 1% absolute reduction in the risk of death, new dialysis, or renal dysfunction lasting to hospital discharge favoring BC.
The SMART and SALT-ED trials have important limitations. They were conducted at a single academic center, and treating clinicians were not blinded to the assigned fluid. The key outcome was a composite of death, new dialysis, and renal dysfunction lasting to hospital discharge – and the trials were not powered to show differences in each of the individual components of the composite.
Despite these limitations, we now have data from two trials enrolling nearly 30,000 acutely ill patients suggesting that BC may result in better clinical outcomes than saline for acutely ill adults. For clinicians who were already using primarily BC solutions, these results will reinforce their current practice. For clinicians whose default IVF has been saline, these new findings raise challenging questions. Prior to these trials, the ICU in which I practice had always used primarily saline. Some of the questions we faced in considering how to apply the results of the SMART and SALT-ED trials to our practice included:
1. Recent data suggest BC may produce better clinical outcomes than saline for acutely ill adults. Are there any data that saline may produce better clinical outcomes than BC? Currently, there are not.
2. Cost is an important consideration in critical care, are BC more expensive than saline? The cost to produce saline and BC is similar. At our hospital, the cost for a 1L bag of saline, LR, and another branded BC (PlasmaLyte A ) is the exactly the same.
3. Is there a specific population for whom BC might have important adverse effects? Because some BC are hypotonic, the safety of administration of BC to patients with elevated intracranial pressure (e.g., traumatic brain injury) is unknown.
4. Are there practical considerations to using BC in the ICU? Compatibility with medications can pose a challenge. For example, the calcium in LR may be incompatible with ceftriaxone infusion. Although BC are compatible with many of the medication infusions used in the ICU for which testing has been performed, less data on compatibility exist for BC than for saline.
5. Are BC as readily available as saline? The three companies that make the majority of IVF used in the United States produce both saline and BC. Recent damage to production facilities has contributed to shortages in the supply of all of them. Over the long term, however, saline and BC are similar in their availability to hospital pharmacies.
After discussing each of these considerations with our ICU physicians and nurses, consultants, and pharmacists, our ICU collectively decided to switch from using primarily saline to BC. This involved (1) our pharmacy team stocking the medication dispensing cabinets in the ICU with 90% LR and 10% saline; and (2) making BC rather than saline the default in order sets within our electronic order entry system. Based on the results of the SMART trial, making the change from saline to BC might be expected to prevent around 100 deaths in our ICU each year.
Many questions regarding the effect of IV crystalloid solutions on clinical outcomes for critically ill adults remain unanswered. The mechanism by which BC may produce better clinical outcomes than saline is uncertain. Whether acetate-containing BC (eg, PlasmaLyte) produced better outcomes than non-acetate-containing BC (eg, LR) is unknown. The safety and efficacy of BC for specific subgroups of patients (eg, those with hyperkalemia) requires further study. Two ongoing trials comparing BC to saline among critically ill adults are expected to finish in 2021 and may provide additional insights into the best approach to IVF management for critically ill adults. An ongoing pilot trial comparing LR to other branded BC (Plasmalyte/Normosol )may inform the choice between BC.
In summary, IVF administration is ubiquitous in critical care. For decades, much of that fluid has been saline. BC are similar to saline in availability and cost. Two large trials now demonstrate better patient outcomes with BC compared with saline. These data challenge ICU providers, pharmacies, and hospital systems primarily using saline to evaluate the available data, their current IVF prescribing practices, and the logistical barriers to change, to determine whether there are legitimate reasons to continue using saline, or whether the time has come to make BC the first-line fluid therapy for acutely ill adults.
Dr. Semler is with the Department of Medicine, Division of Allergy, Pulmonary, and Critical Care Medicine –Vanderbilt University Medical Center, Nashville, Tennessee.
Editor’s Comment
For a very long time, normal saline has been the go-to crystalloid in most ICUs around the globe. In the recent past, evidence started mounting about the potential downside of this solution. The recent SMART trial, the largest to date, indicates that we could prevent adverse renal outcomes by choosing balanced crystalloids over normal saline. These results were even more marked in patients who received a large amount of crystalloids and in patients with sepsis. Dr. Matthew Semler presents solid arguments to consider in changing our practice and adopting a “balanced approach” to fluid resuscitation. We certainly should not only worry about the amount of fluids infused but also about the type of solution we give our patients. Hopefully, we will soon learn if the different balanced solutions also lead to outcome differences.
Angel Coz, MD, FCCP – Section Editor
If you work in an ICU, chances are good that you frequently order IV fluids (IVF). Between resuscitation, maintenance, and medication carriers, nearly all ICU patients receive IVF. Historically, much of this IVF has been 0.9% sodium chloride (“saline” or “normal saline”). Providers in the United States alone administer more than 200 million liters of saline each year (Myburgh JA, et al. N Engl J Med. 2013;369[13]:1243). New evidence, however, suggests that treating your ICU patients with so-called “balanced crystalloids,” rather than saline, may improve patient outcomes.
For over a century, clinicians ordering IV isotonic crystalloids have had two basic options: saline or balanced crystalloids (BC). Saline contains water and 154 mmol/L of sodium chloride (around 50% more chloride than human extracellular fluid). In contrast, BCs, like lactated Ringer’s (LR), Hartman’s solution, and others, contain an amount of chloride resembling human plasma (Table 1). BC substitute an organic anion such as bicarbonate, lactate, acetate, or gluconate, in place of chloride – resulting in lower chloride level and a more neutral pH.
Over the last 2 decades, evidence has slowly accumulated that the different compositions of saline and BC might translate into differences in patient physiology and outcomes. Research in the operating room and ICU found that saline administration caused hyperchloremia and metabolic acidosis. Studies of healthy volunteers found that saline decreased blood flow to the kidney (Chowdhury AH, et al. Ann Surg. 2012;256[1]:18). Animal sepsis models suggested that saline might cause inflammation, low blood pressure, and kidney injury (Zhou F, et al. Crit Care Med. 2014;42[4]:e270). Large observational studies among ICU patients found saline to be associated with increased risk of kidney injury, dialysis, or death (Raghunathan K, et al. Crit Care Med. 2014 Jul;42[7]:1585). These preliminary studies set the stage for a large randomized clinical trial comparing clinical outcomes between BC and saline among acutely ill adults.
Between June 2015 and April 2017, our research group conducted the Isotonic Solutions and Major Adverse Renal Events Trial (SMART) (Semler MW, et al. N Engl J Med. 2018;378[9]:819). SMART was a pragmatic trial in which 15,802 adults in five ICUs were assigned to receive either saline (0.9% sodium chloride) or BC (LR or another branded BC [PlasmaLyte A]). The goal was to determine whether using BC rather than saline would decrease the rates of death, new dialysis, or renal dysfunction lasting through hospital discharge. Patients in the BC group received primarily BC (44% LR and 56% another branded BC [PlasmaLyte A]), whereas patients in the saline group received primarily saline. The rate of death, new dialysis, or renal dysfunction lasting through hospital discharge was lower in the BC group (14.3%) than the saline group (15.4%) (OR: 0.90; 95% CI, 0.82-0.99; P=0.04). The difference between groups was primarily in death and new dialysis, not changes in creatinine. For every 100 patients admitted to an ICU, using BC rather than saline would spare one patient from experiencing death, dialysis, or renal dysfunction lasting to hospital discharge (number needed to treat). The benefits of BC appeared to be greater among patients who received larger volumes of IVF and patients with sepsis. In fact, among patients with sepsis, mortality was significantly lower with BC (25.2%) than with saline (29.4%) (P=.02).
Another trial was conducted in parallel. Saline against LR or another branded BC (PlasmaLyte) in the ED (SALT-ED) compared BC with saline among 13,347 non-critically ill adults treated with IVF in the ED (Self WH, et al. N Engl J Med. 2018;378[9]:829). Like the SMART trial, the SALT-ED trial found a 1% absolute reduction in the risk of death, new dialysis, or renal dysfunction lasting to hospital discharge favoring BC.
The SMART and SALT-ED trials have important limitations. They were conducted at a single academic center, and treating clinicians were not blinded to the assigned fluid. The key outcome was a composite of death, new dialysis, and renal dysfunction lasting to hospital discharge – and the trials were not powered to show differences in each of the individual components of the composite.
Despite these limitations, we now have data from two trials enrolling nearly 30,000 acutely ill patients suggesting that BC may result in better clinical outcomes than saline for acutely ill adults. For clinicians who were already using primarily BC solutions, these results will reinforce their current practice. For clinicians whose default IVF has been saline, these new findings raise challenging questions. Prior to these trials, the ICU in which I practice had always used primarily saline. Some of the questions we faced in considering how to apply the results of the SMART and SALT-ED trials to our practice included:
1. Recent data suggest BC may produce better clinical outcomes than saline for acutely ill adults. Are there any data that saline may produce better clinical outcomes than BC? Currently, there are not.
2. Cost is an important consideration in critical care, are BC more expensive than saline? The cost to produce saline and BC is similar. At our hospital, the cost for a 1L bag of saline, LR, and another branded BC (PlasmaLyte A ) is the exactly the same.
3. Is there a specific population for whom BC might have important adverse effects? Because some BC are hypotonic, the safety of administration of BC to patients with elevated intracranial pressure (e.g., traumatic brain injury) is unknown.
4. Are there practical considerations to using BC in the ICU? Compatibility with medications can pose a challenge. For example, the calcium in LR may be incompatible with ceftriaxone infusion. Although BC are compatible with many of the medication infusions used in the ICU for which testing has been performed, less data on compatibility exist for BC than for saline.
5. Are BC as readily available as saline? The three companies that make the majority of IVF used in the United States produce both saline and BC. Recent damage to production facilities has contributed to shortages in the supply of all of them. Over the long term, however, saline and BC are similar in their availability to hospital pharmacies.
After discussing each of these considerations with our ICU physicians and nurses, consultants, and pharmacists, our ICU collectively decided to switch from using primarily saline to BC. This involved (1) our pharmacy team stocking the medication dispensing cabinets in the ICU with 90% LR and 10% saline; and (2) making BC rather than saline the default in order sets within our electronic order entry system. Based on the results of the SMART trial, making the change from saline to BC might be expected to prevent around 100 deaths in our ICU each year.
Many questions regarding the effect of IV crystalloid solutions on clinical outcomes for critically ill adults remain unanswered. The mechanism by which BC may produce better clinical outcomes than saline is uncertain. Whether acetate-containing BC (eg, PlasmaLyte) produced better outcomes than non-acetate-containing BC (eg, LR) is unknown. The safety and efficacy of BC for specific subgroups of patients (eg, those with hyperkalemia) requires further study. Two ongoing trials comparing BC to saline among critically ill adults are expected to finish in 2021 and may provide additional insights into the best approach to IVF management for critically ill adults. An ongoing pilot trial comparing LR to other branded BC (Plasmalyte/Normosol )may inform the choice between BC.
In summary, IVF administration is ubiquitous in critical care. For decades, much of that fluid has been saline. BC are similar to saline in availability and cost. Two large trials now demonstrate better patient outcomes with BC compared with saline. These data challenge ICU providers, pharmacies, and hospital systems primarily using saline to evaluate the available data, their current IVF prescribing practices, and the logistical barriers to change, to determine whether there are legitimate reasons to continue using saline, or whether the time has come to make BC the first-line fluid therapy for acutely ill adults.
Dr. Semler is with the Department of Medicine, Division of Allergy, Pulmonary, and Critical Care Medicine –Vanderbilt University Medical Center, Nashville, Tennessee.
Editor’s Comment
For a very long time, normal saline has been the go-to crystalloid in most ICUs around the globe. In the recent past, evidence started mounting about the potential downside of this solution. The recent SMART trial, the largest to date, indicates that we could prevent adverse renal outcomes by choosing balanced crystalloids over normal saline. These results were even more marked in patients who received a large amount of crystalloids and in patients with sepsis. Dr. Matthew Semler presents solid arguments to consider in changing our practice and adopting a “balanced approach” to fluid resuscitation. We certainly should not only worry about the amount of fluids infused but also about the type of solution we give our patients. Hopefully, we will soon learn if the different balanced solutions also lead to outcome differences.
Angel Coz, MD, FCCP – Section Editor
If you work in an ICU, chances are good that you frequently order IV fluids (IVF). Between resuscitation, maintenance, and medication carriers, nearly all ICU patients receive IVF. Historically, much of this IVF has been 0.9% sodium chloride (“saline” or “normal saline”). Providers in the United States alone administer more than 200 million liters of saline each year (Myburgh JA, et al. N Engl J Med. 2013;369[13]:1243). New evidence, however, suggests that treating your ICU patients with so-called “balanced crystalloids,” rather than saline, may improve patient outcomes.
For over a century, clinicians ordering IV isotonic crystalloids have had two basic options: saline or balanced crystalloids (BC). Saline contains water and 154 mmol/L of sodium chloride (around 50% more chloride than human extracellular fluid). In contrast, BCs, like lactated Ringer’s (LR), Hartman’s solution, and others, contain an amount of chloride resembling human plasma (Table 1). BC substitute an organic anion such as bicarbonate, lactate, acetate, or gluconate, in place of chloride – resulting in lower chloride level and a more neutral pH.
Over the last 2 decades, evidence has slowly accumulated that the different compositions of saline and BC might translate into differences in patient physiology and outcomes. Research in the operating room and ICU found that saline administration caused hyperchloremia and metabolic acidosis. Studies of healthy volunteers found that saline decreased blood flow to the kidney (Chowdhury AH, et al. Ann Surg. 2012;256[1]:18). Animal sepsis models suggested that saline might cause inflammation, low blood pressure, and kidney injury (Zhou F, et al. Crit Care Med. 2014;42[4]:e270). Large observational studies among ICU patients found saline to be associated with increased risk of kidney injury, dialysis, or death (Raghunathan K, et al. Crit Care Med. 2014 Jul;42[7]:1585). These preliminary studies set the stage for a large randomized clinical trial comparing clinical outcomes between BC and saline among acutely ill adults.
Between June 2015 and April 2017, our research group conducted the Isotonic Solutions and Major Adverse Renal Events Trial (SMART) (Semler MW, et al. N Engl J Med. 2018;378[9]:819). SMART was a pragmatic trial in which 15,802 adults in five ICUs were assigned to receive either saline (0.9% sodium chloride) or BC (LR or another branded BC [PlasmaLyte A]). The goal was to determine whether using BC rather than saline would decrease the rates of death, new dialysis, or renal dysfunction lasting through hospital discharge. Patients in the BC group received primarily BC (44% LR and 56% another branded BC [PlasmaLyte A]), whereas patients in the saline group received primarily saline. The rate of death, new dialysis, or renal dysfunction lasting through hospital discharge was lower in the BC group (14.3%) than the saline group (15.4%) (OR: 0.90; 95% CI, 0.82-0.99; P=0.04). The difference between groups was primarily in death and new dialysis, not changes in creatinine. For every 100 patients admitted to an ICU, using BC rather than saline would spare one patient from experiencing death, dialysis, or renal dysfunction lasting to hospital discharge (number needed to treat). The benefits of BC appeared to be greater among patients who received larger volumes of IVF and patients with sepsis. In fact, among patients with sepsis, mortality was significantly lower with BC (25.2%) than with saline (29.4%) (P=.02).
Another trial was conducted in parallel. Saline against LR or another branded BC (PlasmaLyte) in the ED (SALT-ED) compared BC with saline among 13,347 non-critically ill adults treated with IVF in the ED (Self WH, et al. N Engl J Med. 2018;378[9]:829). Like the SMART trial, the SALT-ED trial found a 1% absolute reduction in the risk of death, new dialysis, or renal dysfunction lasting to hospital discharge favoring BC.
The SMART and SALT-ED trials have important limitations. They were conducted at a single academic center, and treating clinicians were not blinded to the assigned fluid. The key outcome was a composite of death, new dialysis, and renal dysfunction lasting to hospital discharge – and the trials were not powered to show differences in each of the individual components of the composite.
Despite these limitations, we now have data from two trials enrolling nearly 30,000 acutely ill patients suggesting that BC may result in better clinical outcomes than saline for acutely ill adults. For clinicians who were already using primarily BC solutions, these results will reinforce their current practice. For clinicians whose default IVF has been saline, these new findings raise challenging questions. Prior to these trials, the ICU in which I practice had always used primarily saline. Some of the questions we faced in considering how to apply the results of the SMART and SALT-ED trials to our practice included:
1. Recent data suggest BC may produce better clinical outcomes than saline for acutely ill adults. Are there any data that saline may produce better clinical outcomes than BC? Currently, there are not.
2. Cost is an important consideration in critical care, are BC more expensive than saline? The cost to produce saline and BC is similar. At our hospital, the cost for a 1L bag of saline, LR, and another branded BC (PlasmaLyte A ) is the exactly the same.
3. Is there a specific population for whom BC might have important adverse effects? Because some BC are hypotonic, the safety of administration of BC to patients with elevated intracranial pressure (e.g., traumatic brain injury) is unknown.
4. Are there practical considerations to using BC in the ICU? Compatibility with medications can pose a challenge. For example, the calcium in LR may be incompatible with ceftriaxone infusion. Although BC are compatible with many of the medication infusions used in the ICU for which testing has been performed, less data on compatibility exist for BC than for saline.
5. Are BC as readily available as saline? The three companies that make the majority of IVF used in the United States produce both saline and BC. Recent damage to production facilities has contributed to shortages in the supply of all of them. Over the long term, however, saline and BC are similar in their availability to hospital pharmacies.
After discussing each of these considerations with our ICU physicians and nurses, consultants, and pharmacists, our ICU collectively decided to switch from using primarily saline to BC. This involved (1) our pharmacy team stocking the medication dispensing cabinets in the ICU with 90% LR and 10% saline; and (2) making BC rather than saline the default in order sets within our electronic order entry system. Based on the results of the SMART trial, making the change from saline to BC might be expected to prevent around 100 deaths in our ICU each year.
Many questions regarding the effect of IV crystalloid solutions on clinical outcomes for critically ill adults remain unanswered. The mechanism by which BC may produce better clinical outcomes than saline is uncertain. Whether acetate-containing BC (eg, PlasmaLyte) produced better outcomes than non-acetate-containing BC (eg, LR) is unknown. The safety and efficacy of BC for specific subgroups of patients (eg, those with hyperkalemia) requires further study. Two ongoing trials comparing BC to saline among critically ill adults are expected to finish in 2021 and may provide additional insights into the best approach to IVF management for critically ill adults. An ongoing pilot trial comparing LR to other branded BC (Plasmalyte/Normosol )may inform the choice between BC.
In summary, IVF administration is ubiquitous in critical care. For decades, much of that fluid has been saline. BC are similar to saline in availability and cost. Two large trials now demonstrate better patient outcomes with BC compared with saline. These data challenge ICU providers, pharmacies, and hospital systems primarily using saline to evaluate the available data, their current IVF prescribing practices, and the logistical barriers to change, to determine whether there are legitimate reasons to continue using saline, or whether the time has come to make BC the first-line fluid therapy for acutely ill adults.
Dr. Semler is with the Department of Medicine, Division of Allergy, Pulmonary, and Critical Care Medicine –Vanderbilt University Medical Center, Nashville, Tennessee.
Editor’s Comment
For a very long time, normal saline has been the go-to crystalloid in most ICUs around the globe. In the recent past, evidence started mounting about the potential downside of this solution. The recent SMART trial, the largest to date, indicates that we could prevent adverse renal outcomes by choosing balanced crystalloids over normal saline. These results were even more marked in patients who received a large amount of crystalloids and in patients with sepsis. Dr. Matthew Semler presents solid arguments to consider in changing our practice and adopting a “balanced approach” to fluid resuscitation. We certainly should not only worry about the amount of fluids infused but also about the type of solution we give our patients. Hopefully, we will soon learn if the different balanced solutions also lead to outcome differences.
Angel Coz, MD, FCCP – Section Editor
Diagnosis and Management of Critical Illness-Related Corticosteroid Insufficiency (CIRCI): Updated Guidelines 2017
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines

Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines
Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines
Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Life after angiotensin II
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.

Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).

Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.
Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).
Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.
Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).
Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
On Diagnosing Sepsis
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.

However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.
However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.
However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Clostridium difficile in the ICU: A “fluid” issue
In critically ill patients admitted to the ICU, diarrhea (defined as three or more watery loose stools within 24 hours) is a common problem. The etiologies of diarrhea are many, with infectious and noninfectious causes encountered.
Clostridium difficile infection (CDI) is the most common infectious cause of diarrhea in the hospital, including the ICU. The Centers for Disease Control and Prevention estimates the number of overall CDI cases to number about a half-million per year, of which 1 in 5 patients will have a recurrence, and 1 in 11 people aged ≥65 years will die within a month of CDI diagnosis. Age is a poor prognostic risk; greater than 80% of C difficile deaths occur in people 65 and older.
The increased use of electronic sepsis screening tools and aggressive antibiotic treatment, often done through protocols, has recently been identified as paradoxically increasing CDI occurrence (Hiensch R et al. Am J Infect Control. 2017;45[10]:1091). However, similar rapid identification and management of CDI can result in improved patient outcomes.
Issues with diagnosing CDI
Episodes of CDI can be rapid and severe, especially if due to hyper-toxin producing–strains of C difficile, such as BI/NAP1/027, which produces significantly higher levels of Toxin A, Toxin B, and binary toxin CDT (Denève C, et al. Int J Antimicrob Agents. 2009;33:S24). Testing for CDI has been controversial; several methods have been employed to aid in the diagnosis of CDI. Currently, many institutions use either nucleic acid amplification tests (NAATs) for toxigenic C difficile or direct detection of the toxin produced by the bacteria. NAATs and past culture-based methods are more sensitive but less specific than toxin assays, whereas toxin assays are less sensitive but more specific than NAATs. However, detection of C difficile colonization due to high-sensitivity NAATs has caused a rise in the apparent rate of hospital-acquired CDI (Polage CR, et al. JAMA Intern Med. 2015;175[11]:4114).
To counter this, multi-step algorithmic approaches to CDI diagnosis have been recommended, including the use of glutamate dehydrogenase (GDH) antigen, toxin detection, and NAATs for toxin-producing C difficile. These multistep pathways attempt to minimize false-positive test results while affirming the presence or absence of true CDI (Fang F, et al. J Clin Microbiol. 2017; 55[3]:670).
However, controversy continues regarding which testing modalities are optimal, as some patients with positive toxin assays have asymptomatic colonization while some patients with negative toxin assays have CDI. The hope is that emerging, higher sensitivity toxin assays will decrease the number of CDI cases missed by negative toxin tests. Because C difficile toxins are labile at body temperature and susceptible to inactivation by digestive enzymes, stool samples must be expeditiously transported to the lab (time is of the essence), so as not to lose toxin or NAAT target detection. Repeat CDI testing for a “test for cure” is not recommended.
Management of CDI
The initial management of CDI has been discussed in many publications, including the current SHEA/IDSA Guidelines (Cohen SH, et al. Infect Control Hosp Epidemiol. 2010;31[5]:431).
Briefly, this involves stratifying CDI patients by clinical severity (mild, moderate, severe) and objective data (leukocytosis >15,000, septic shock, serum creatinine level > 1.5 times premorbid level) to guide initial antibiotic therapy. For mild/moderate first episode of CDI, oral or IV metronidazole is generally recommended; more severe disease is generally treated with oral vancomycin.
Complicated CDI in patients (hypotension/shock, ileus, toxic megacolon) requires aggressive management with both IV metronidazole and oral vancomycin (if ileus is present, consider vancomycin enemas). Additionally, fidaxomicin is available for oral CDI treatment and has been associated with decreased first-episode CDI recurrence.
The management of CDI recurrence commonly involves using oral vancomycin as a taper (or taper/pulse regimen) or using fidaxomicin. A recent publication (Sirbu et al. Clin Infect Dis. 2017;65[8]:1396) retrospectively compared vancomycin taper and pulse treatment strategies for 100 consecutive patients with CDI.
After taper, patents who received every other day (QOD) dosing had a cure rate of 61%, while those who received QOD dosing followed by every third day dosing achieved an 81% cure rate. A clinical trial comparing vancomycin standard therapy vs vancomycin taper with pulse vs fidaxomicin for first- and second-recurrence of CDI is underway.
Last year, the FDA approved bezlotoxumab, a monoclonal antibody that binds to C difficile toxin B. Bezlotoxumab treatment is indicated to reduce CDI recurrence in patients >18 years of age and is administered while CDI antibiotic therapy is ongoing.
When comparing 12-week efficacy using standard of care (SoC) CDI treatment vs SoC plus bezlotoxumab (SoC+Bmab), recurrence rates in SoC and SoC+Bmab were 27.6% vs1 7.4%, respectively, in one trial, and 25.7% vs 15.7% in another. While generally well-tolerated, bezlotoxumab is associated with increased risk for exacerbating heart failure. Data relating to the cost-effectiveness of bezlotoxumab are currently pending.
Fecal microbiota transplant (FMT)— duodenal or colonic instillation of donor fecal microbiota to “restore” normal flora— is an evolving CDI therapy with promising results but difficult administration. Although FMT has high published success rates, the FDA’s policy of “enforcement discretion” permits practitioners to proceed with FMT only as an Investigational New Drug. This requires signed, informed consent to FMT as an investigational therapy with unknown long-term risks.
The FDA deemed these protections necessary as ongoing studies of the human microbiome have yet to define what constitutes “normal flora,” and some investigators highlight the possibility of transmitting flora or gut factors associated with obesity, metabolic syndrome, or malignancy.
Experimental CDI preventive modalities include new antibiotics, monoclonal antibodies, probiotics, select other novel agents, and C. difficile vaccinations. These vaccines include recombinant fusion proteins and adjuvant toxoids, both of which have generally favorable tolerance profiles, as well as robust immune responses in clinical trial subjects. However, the efficacy of these vaccines at preventing clinical disease is still to be demonstrated.
Lastly, the ubiquitous use of proton pump inhibitors (PPI) in ICUs plays a role in promoting CDI incidence, severity, and recurrence. Accordingly, the pros and cons of PPI use must be weighed in each patient.
CDI prevention in the hospital environment
Hospital-acquired CDIs (HA-CDI) and nosocomial transmission clearly occur. A recent study of electronic health record data demonstrated that patients who passed through the hospital’s emergency department CT scanner within 24 hours after a patient with C difficile were twice as likely to become infected (Murray SG, et al. JAMA Internal Medicine. published online October 23, 2017. doi:10.1001). Receipt of antibiotics by prior bed occupants was associated with increased risk for CDI in subsequent patients, implying that antibiotics can directly affect the risk for CDI in patients who do not themselves receive antibiotics. As such, aggressive environmental cleaning in conjunction with hospital antimicrobial stewardship efforts, such as appropriate use of antibiotics known to increase CDI occurrence, are required to minimize HA-CDI.
Contact precautions should be strictly enforced; wearing gloves and gowns is necessary for every encounter when treating patients with C difficile, even during short visits. Hand sanitizer does not kill C difficile, and although soap-and-water hand washing works better, it may be insufficient alone, reinforcing the importance of using gloves with all patient encounters.
The strain placed on ICUs by CDI has been increasing over the past several years. Physicians and hospitals are at risk for lower performance scores and reduced reimbursement due to CDI relapses. As such, burgeoning areas of debate and research include efforts to quickly and accurately diagnose CDI along with reducing recurrence rates. Yet, with all the capital investment, the most significant and cost-effective method to reduce CDI rates remains proper and frequent hand washing with soap and water. Prevention of disease remains the cornerstone to treatment.
In critically ill patients admitted to the ICU, diarrhea (defined as three or more watery loose stools within 24 hours) is a common problem. The etiologies of diarrhea are many, with infectious and noninfectious causes encountered.
Clostridium difficile infection (CDI) is the most common infectious cause of diarrhea in the hospital, including the ICU. The Centers for Disease Control and Prevention estimates the number of overall CDI cases to number about a half-million per year, of which 1 in 5 patients will have a recurrence, and 1 in 11 people aged ≥65 years will die within a month of CDI diagnosis. Age is a poor prognostic risk; greater than 80% of C difficile deaths occur in people 65 and older.
The increased use of electronic sepsis screening tools and aggressive antibiotic treatment, often done through protocols, has recently been identified as paradoxically increasing CDI occurrence (Hiensch R et al. Am J Infect Control. 2017;45[10]:1091). However, similar rapid identification and management of CDI can result in improved patient outcomes.
Issues with diagnosing CDI
Episodes of CDI can be rapid and severe, especially if due to hyper-toxin producing–strains of C difficile, such as BI/NAP1/027, which produces significantly higher levels of Toxin A, Toxin B, and binary toxin CDT (Denève C, et al. Int J Antimicrob Agents. 2009;33:S24). Testing for CDI has been controversial; several methods have been employed to aid in the diagnosis of CDI. Currently, many institutions use either nucleic acid amplification tests (NAATs) for toxigenic C difficile or direct detection of the toxin produced by the bacteria. NAATs and past culture-based methods are more sensitive but less specific than toxin assays, whereas toxin assays are less sensitive but more specific than NAATs. However, detection of C difficile colonization due to high-sensitivity NAATs has caused a rise in the apparent rate of hospital-acquired CDI (Polage CR, et al. JAMA Intern Med. 2015;175[11]:4114).
To counter this, multi-step algorithmic approaches to CDI diagnosis have been recommended, including the use of glutamate dehydrogenase (GDH) antigen, toxin detection, and NAATs for toxin-producing C difficile. These multistep pathways attempt to minimize false-positive test results while affirming the presence or absence of true CDI (Fang F, et al. J Clin Microbiol. 2017; 55[3]:670).
However, controversy continues regarding which testing modalities are optimal, as some patients with positive toxin assays have asymptomatic colonization while some patients with negative toxin assays have CDI. The hope is that emerging, higher sensitivity toxin assays will decrease the number of CDI cases missed by negative toxin tests. Because C difficile toxins are labile at body temperature and susceptible to inactivation by digestive enzymes, stool samples must be expeditiously transported to the lab (time is of the essence), so as not to lose toxin or NAAT target detection. Repeat CDI testing for a “test for cure” is not recommended.
Management of CDI
The initial management of CDI has been discussed in many publications, including the current SHEA/IDSA Guidelines (Cohen SH, et al. Infect Control Hosp Epidemiol. 2010;31[5]:431).
Briefly, this involves stratifying CDI patients by clinical severity (mild, moderate, severe) and objective data (leukocytosis >15,000, septic shock, serum creatinine level > 1.5 times premorbid level) to guide initial antibiotic therapy. For mild/moderate first episode of CDI, oral or IV metronidazole is generally recommended; more severe disease is generally treated with oral vancomycin.
Complicated CDI in patients (hypotension/shock, ileus, toxic megacolon) requires aggressive management with both IV metronidazole and oral vancomycin (if ileus is present, consider vancomycin enemas). Additionally, fidaxomicin is available for oral CDI treatment and has been associated with decreased first-episode CDI recurrence.
The management of CDI recurrence commonly involves using oral vancomycin as a taper (or taper/pulse regimen) or using fidaxomicin. A recent publication (Sirbu et al. Clin Infect Dis. 2017;65[8]:1396) retrospectively compared vancomycin taper and pulse treatment strategies for 100 consecutive patients with CDI.
After taper, patents who received every other day (QOD) dosing had a cure rate of 61%, while those who received QOD dosing followed by every third day dosing achieved an 81% cure rate. A clinical trial comparing vancomycin standard therapy vs vancomycin taper with pulse vs fidaxomicin for first- and second-recurrence of CDI is underway.
Last year, the FDA approved bezlotoxumab, a monoclonal antibody that binds to C difficile toxin B. Bezlotoxumab treatment is indicated to reduce CDI recurrence in patients >18 years of age and is administered while CDI antibiotic therapy is ongoing.
When comparing 12-week efficacy using standard of care (SoC) CDI treatment vs SoC plus bezlotoxumab (SoC+Bmab), recurrence rates in SoC and SoC+Bmab were 27.6% vs1 7.4%, respectively, in one trial, and 25.7% vs 15.7% in another. While generally well-tolerated, bezlotoxumab is associated with increased risk for exacerbating heart failure. Data relating to the cost-effectiveness of bezlotoxumab are currently pending.
Fecal microbiota transplant (FMT)— duodenal or colonic instillation of donor fecal microbiota to “restore” normal flora— is an evolving CDI therapy with promising results but difficult administration. Although FMT has high published success rates, the FDA’s policy of “enforcement discretion” permits practitioners to proceed with FMT only as an Investigational New Drug. This requires signed, informed consent to FMT as an investigational therapy with unknown long-term risks.
The FDA deemed these protections necessary as ongoing studies of the human microbiome have yet to define what constitutes “normal flora,” and some investigators highlight the possibility of transmitting flora or gut factors associated with obesity, metabolic syndrome, or malignancy.
Experimental CDI preventive modalities include new antibiotics, monoclonal antibodies, probiotics, select other novel agents, and C. difficile vaccinations. These vaccines include recombinant fusion proteins and adjuvant toxoids, both of which have generally favorable tolerance profiles, as well as robust immune responses in clinical trial subjects. However, the efficacy of these vaccines at preventing clinical disease is still to be demonstrated.
Lastly, the ubiquitous use of proton pump inhibitors (PPI) in ICUs plays a role in promoting CDI incidence, severity, and recurrence. Accordingly, the pros and cons of PPI use must be weighed in each patient.
CDI prevention in the hospital environment
Hospital-acquired CDIs (HA-CDI) and nosocomial transmission clearly occur. A recent study of electronic health record data demonstrated that patients who passed through the hospital’s emergency department CT scanner within 24 hours after a patient with C difficile were twice as likely to become infected (Murray SG, et al. JAMA Internal Medicine. published online October 23, 2017. doi:10.1001). Receipt of antibiotics by prior bed occupants was associated with increased risk for CDI in subsequent patients, implying that antibiotics can directly affect the risk for CDI in patients who do not themselves receive antibiotics. As such, aggressive environmental cleaning in conjunction with hospital antimicrobial stewardship efforts, such as appropriate use of antibiotics known to increase CDI occurrence, are required to minimize HA-CDI.
Contact precautions should be strictly enforced; wearing gloves and gowns is necessary for every encounter when treating patients with C difficile, even during short visits. Hand sanitizer does not kill C difficile, and although soap-and-water hand washing works better, it may be insufficient alone, reinforcing the importance of using gloves with all patient encounters.
The strain placed on ICUs by CDI has been increasing over the past several years. Physicians and hospitals are at risk for lower performance scores and reduced reimbursement due to CDI relapses. As such, burgeoning areas of debate and research include efforts to quickly and accurately diagnose CDI along with reducing recurrence rates. Yet, with all the capital investment, the most significant and cost-effective method to reduce CDI rates remains proper and frequent hand washing with soap and water. Prevention of disease remains the cornerstone to treatment.
In critically ill patients admitted to the ICU, diarrhea (defined as three or more watery loose stools within 24 hours) is a common problem. The etiologies of diarrhea are many, with infectious and noninfectious causes encountered.
Clostridium difficile infection (CDI) is the most common infectious cause of diarrhea in the hospital, including the ICU. The Centers for Disease Control and Prevention estimates the number of overall CDI cases to number about a half-million per year, of which 1 in 5 patients will have a recurrence, and 1 in 11 people aged ≥65 years will die within a month of CDI diagnosis. Age is a poor prognostic risk; greater than 80% of C difficile deaths occur in people 65 and older.
The increased use of electronic sepsis screening tools and aggressive antibiotic treatment, often done through protocols, has recently been identified as paradoxically increasing CDI occurrence (Hiensch R et al. Am J Infect Control. 2017;45[10]:1091). However, similar rapid identification and management of CDI can result in improved patient outcomes.
Issues with diagnosing CDI
Episodes of CDI can be rapid and severe, especially if due to hyper-toxin producing–strains of C difficile, such as BI/NAP1/027, which produces significantly higher levels of Toxin A, Toxin B, and binary toxin CDT (Denève C, et al. Int J Antimicrob Agents. 2009;33:S24). Testing for CDI has been controversial; several methods have been employed to aid in the diagnosis of CDI. Currently, many institutions use either nucleic acid amplification tests (NAATs) for toxigenic C difficile or direct detection of the toxin produced by the bacteria. NAATs and past culture-based methods are more sensitive but less specific than toxin assays, whereas toxin assays are less sensitive but more specific than NAATs. However, detection of C difficile colonization due to high-sensitivity NAATs has caused a rise in the apparent rate of hospital-acquired CDI (Polage CR, et al. JAMA Intern Med. 2015;175[11]:4114).
To counter this, multi-step algorithmic approaches to CDI diagnosis have been recommended, including the use of glutamate dehydrogenase (GDH) antigen, toxin detection, and NAATs for toxin-producing C difficile. These multistep pathways attempt to minimize false-positive test results while affirming the presence or absence of true CDI (Fang F, et al. J Clin Microbiol. 2017; 55[3]:670).
However, controversy continues regarding which testing modalities are optimal, as some patients with positive toxin assays have asymptomatic colonization while some patients with negative toxin assays have CDI. The hope is that emerging, higher sensitivity toxin assays will decrease the number of CDI cases missed by negative toxin tests. Because C difficile toxins are labile at body temperature and susceptible to inactivation by digestive enzymes, stool samples must be expeditiously transported to the lab (time is of the essence), so as not to lose toxin or NAAT target detection. Repeat CDI testing for a “test for cure” is not recommended.
Management of CDI
The initial management of CDI has been discussed in many publications, including the current SHEA/IDSA Guidelines (Cohen SH, et al. Infect Control Hosp Epidemiol. 2010;31[5]:431).
Briefly, this involves stratifying CDI patients by clinical severity (mild, moderate, severe) and objective data (leukocytosis >15,000, septic shock, serum creatinine level > 1.5 times premorbid level) to guide initial antibiotic therapy. For mild/moderate first episode of CDI, oral or IV metronidazole is generally recommended; more severe disease is generally treated with oral vancomycin.
Complicated CDI in patients (hypotension/shock, ileus, toxic megacolon) requires aggressive management with both IV metronidazole and oral vancomycin (if ileus is present, consider vancomycin enemas). Additionally, fidaxomicin is available for oral CDI treatment and has been associated with decreased first-episode CDI recurrence.
The management of CDI recurrence commonly involves using oral vancomycin as a taper (or taper/pulse regimen) or using fidaxomicin. A recent publication (Sirbu et al. Clin Infect Dis. 2017;65[8]:1396) retrospectively compared vancomycin taper and pulse treatment strategies for 100 consecutive patients with CDI.
After taper, patents who received every other day (QOD) dosing had a cure rate of 61%, while those who received QOD dosing followed by every third day dosing achieved an 81% cure rate. A clinical trial comparing vancomycin standard therapy vs vancomycin taper with pulse vs fidaxomicin for first- and second-recurrence of CDI is underway.
Last year, the FDA approved bezlotoxumab, a monoclonal antibody that binds to C difficile toxin B. Bezlotoxumab treatment is indicated to reduce CDI recurrence in patients >18 years of age and is administered while CDI antibiotic therapy is ongoing.
When comparing 12-week efficacy using standard of care (SoC) CDI treatment vs SoC plus bezlotoxumab (SoC+Bmab), recurrence rates in SoC and SoC+Bmab were 27.6% vs1 7.4%, respectively, in one trial, and 25.7% vs 15.7% in another. While generally well-tolerated, bezlotoxumab is associated with increased risk for exacerbating heart failure. Data relating to the cost-effectiveness of bezlotoxumab are currently pending.
Fecal microbiota transplant (FMT)— duodenal or colonic instillation of donor fecal microbiota to “restore” normal flora— is an evolving CDI therapy with promising results but difficult administration. Although FMT has high published success rates, the FDA’s policy of “enforcement discretion” permits practitioners to proceed with FMT only as an Investigational New Drug. This requires signed, informed consent to FMT as an investigational therapy with unknown long-term risks.
The FDA deemed these protections necessary as ongoing studies of the human microbiome have yet to define what constitutes “normal flora,” and some investigators highlight the possibility of transmitting flora or gut factors associated with obesity, metabolic syndrome, or malignancy.
Experimental CDI preventive modalities include new antibiotics, monoclonal antibodies, probiotics, select other novel agents, and C. difficile vaccinations. These vaccines include recombinant fusion proteins and adjuvant toxoids, both of which have generally favorable tolerance profiles, as well as robust immune responses in clinical trial subjects. However, the efficacy of these vaccines at preventing clinical disease is still to be demonstrated.
Lastly, the ubiquitous use of proton pump inhibitors (PPI) in ICUs plays a role in promoting CDI incidence, severity, and recurrence. Accordingly, the pros and cons of PPI use must be weighed in each patient.
CDI prevention in the hospital environment
Hospital-acquired CDIs (HA-CDI) and nosocomial transmission clearly occur. A recent study of electronic health record data demonstrated that patients who passed through the hospital’s emergency department CT scanner within 24 hours after a patient with C difficile were twice as likely to become infected (Murray SG, et al. JAMA Internal Medicine. published online October 23, 2017. doi:10.1001). Receipt of antibiotics by prior bed occupants was associated with increased risk for CDI in subsequent patients, implying that antibiotics can directly affect the risk for CDI in patients who do not themselves receive antibiotics. As such, aggressive environmental cleaning in conjunction with hospital antimicrobial stewardship efforts, such as appropriate use of antibiotics known to increase CDI occurrence, are required to minimize HA-CDI.
Contact precautions should be strictly enforced; wearing gloves and gowns is necessary for every encounter when treating patients with C difficile, even during short visits. Hand sanitizer does not kill C difficile, and although soap-and-water hand washing works better, it may be insufficient alone, reinforcing the importance of using gloves with all patient encounters.
The strain placed on ICUs by CDI has been increasing over the past several years. Physicians and hospitals are at risk for lower performance scores and reduced reimbursement due to CDI relapses. As such, burgeoning areas of debate and research include efforts to quickly and accurately diagnose CDI along with reducing recurrence rates. Yet, with all the capital investment, the most significant and cost-effective method to reduce CDI rates remains proper and frequent hand washing with soap and water. Prevention of disease remains the cornerstone to treatment.