User login
Intermittent pain and stiffness
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are consistent with a diagnosis of psoriatic spondylitis.
Psoriatic spondylitis is a form of psoriatic arthritis (PsA) that affects the spine and the joints in the pelvis (axial involvement). PsA is a chronic, heterogeneous condition that affects approximately 25%-30% of patients with psoriasis, particularly those with severe psoriasis or nail or scalp involvement. It is characterized by musculoskeletal inflammation (arthritis, enthesitis, spondylitis, and dactylitis). PsA is a spondyloarthritis that can be found either in the peripheral or axial skeleton. If not treated, it may result in permanent joint damage and loss of function.
Patients with PsA may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Common symptoms of axial involvement in PsA include morning back/neck stiffness that lasts longer than 30 minutes, neck or back pain that improves with activity and worsens after prolonged inactivity, and diminished mobility. PsA affects men and women equally, and typically develops when patients are between 30 and 50 years of age. As with psoriasis, PsA is associated with numerous comorbidities, such as cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
The diagnosis of psoriatic spondylitis is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosus). Useful imaging tools for evaluating patients with PsA include plain radiography, CT, ultrasound, and MRI. Although MRI and ultrasound may be more sensitive than plain radiography for detecting early joint inflammation and damage and axial changes, including sacroiliitis, they are not mandatory for a diagnosis of PsA to be made.
International guidelines have been developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA), the European Alliance of Associations for Rheumatology (EULAR), and the Assessment of Spondyloarthritis International Society to guide the treatment of axial PsA. The goals of treatment include minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; improving functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Treatment options for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or have erosive disease or other indications of high disease activity, guidelines recommend initiation of a tumor necrosis factor (TNF) inhibitor (eg, adalimumab, etanercept, infliximab, golimumab, certolizumab pegol). Disease-modifying antirheumatic drugs (eg, methotrexate) are not routinely prescribed for patients with axial disease because they have not been shown to be effective. In patients with significant skin involvement, treatment with interleukin-17A inhibitors may be preferred to TNF inhibitors.
If patients have an inadequate response to a first trial of a TNF inhibitor, guidelines recommend trying a second TNF inhibitor before switching to a different class of biologic. For patients who do not respond to TNF inhibitors, a Janus kinase inhibitor (tofacitinib) may be considered. Additionally, nonpharmacologic therapies (eg, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 41-year-old man with a 5-year history of moderate to severe scalp psoriasis presents with complaints of intermittent pain and stiffness in his left hip and lower back of approximately 6 months' duration. The patient states that his back pain has been severe enough to wake him up on several occasions. Treatment with over-the-counter ibuprofen is moderately effective at relieving his pain. He also reports morning back stiffness that improves with motion, usually within an hour of awakening. The patient reports no fever, pain, swelling, or worsening of his scalp psoriasis. He is not aware of any injury or other triggering factor for his back pain. He takes an over-the-counter multivitamin daily and treats his scalp psoriasis with fluocinolone acetonide 0.01% oil. The patient is 5 ft 9 in and weighs 176 lb (BMI 26).
Physical examination reveals tenderness in the lumbar spine and associated decreased range of motion, as well as psoriatic plaques on the scalp. Vital signs are within normal ranges. Pertinent laboratory findings include erythrocyte sedimentation rate of 19 mm/h and C-reactive protein of 10 mg/L. Rheumatoid factor, antinuclear antibody, and anti-cyclic citrullinated peptide antibody were negative. Radiographic findings include sacroiliitis and bulky nonmarginal syndesmophytes.
Skin changes and pain
The history and findings in this case are suggestive of inflammatory breast cancer.
Breast cancer is the leading life-threatening cancer diagnosed and the second-leading cause of cancer-related deaths in women worldwide. In the United States, estimates suggest that 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 women died of the disease. Globally, approximately 2.3 million new diagnoses and 685,000 breast cancer–related deaths were reported in 2020.
Inflammatory breast cancer is a rare and highly aggressive subtype of locally advanced breast cancer. In the United States, inflammatory breast cancer accounts for approximately 2%-4% of breast cancer cases. Although its incidence is rare, 7% of breast cancer caused mortality is attributed to inflammatory breast cancer. Cases of inflammatory breast cancer tend to be diagnosed at a younger age compared with noninflammatory breast cancer cases. Risk factors include African-American race and obesity.
The symptoms of inflammatory breast cancer can vary broadly, ranging from subtle skin erythema to diffuse breast involvement with skin dimpling and nipple inversion. Diagnostic criteria include erythema occupying at least one third of the breast, edema, peau d'orange, and/or warmth, with or without an underlying mass; rapid onset (< 3 months); and pathologic confirmation of invasive breast carcinoma. Histologic findings include florid tumor emboli that obstruct dermal lymphatics, which results in swelling and inflammation of the affected breast.
Inflammatory breast cancer has been associated with a poor prognosis. However, treatment advances are helping to improve outcomes. Currently, 5-year survival rates are reported to be 40%-70%, with a median survival of 2-4 years. According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the first-line treatment of inflammatory breast cancer involves neoadjuvant chemotherapy, modified radical mastectomy, and adjuvant radiation to the chest wall and regional nodes. Endocrine treatment should also be given to patients who are ER-positive and/or PR-positive (sequential chemotherapy followed by endocrine therapy). For patients who are HER2-positive, up to 1 year of HER2-targeted therapy should be given. HER2-targeted therapies can be administered concurrently with radiation and with endocrine therapy if indicated.
Delayed reconstruction after mastectomy remains the clinical standard for inflammatory breast cancer. This is because the need to resect involved skin negates the benefit of skin-sparing mastectomy for immediate reconstruction. Moreover, high rates of local and distant recurrence warrant comprehensive regional node irradiation in a timely fashion, which may be more challenging or subject to delay after immediate reconstruction. Rarely, the extent of skin excision at the time of mastectomy prohibits primary or local closure. In such cases, reconstruction of the chest wall defect with autologous tissue is required, and concomitant immediate reconstruction may be undertaken.
Detailed guidance on the treatment of inflammatory breast cancer, in the first line and beyond, are available from the NCCN.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of inflammatory breast cancer.
Breast cancer is the leading life-threatening cancer diagnosed and the second-leading cause of cancer-related deaths in women worldwide. In the United States, estimates suggest that 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 women died of the disease. Globally, approximately 2.3 million new diagnoses and 685,000 breast cancer–related deaths were reported in 2020.
Inflammatory breast cancer is a rare and highly aggressive subtype of locally advanced breast cancer. In the United States, inflammatory breast cancer accounts for approximately 2%-4% of breast cancer cases. Although its incidence is rare, 7% of breast cancer caused mortality is attributed to inflammatory breast cancer. Cases of inflammatory breast cancer tend to be diagnosed at a younger age compared with noninflammatory breast cancer cases. Risk factors include African-American race and obesity.
The symptoms of inflammatory breast cancer can vary broadly, ranging from subtle skin erythema to diffuse breast involvement with skin dimpling and nipple inversion. Diagnostic criteria include erythema occupying at least one third of the breast, edema, peau d'orange, and/or warmth, with or without an underlying mass; rapid onset (< 3 months); and pathologic confirmation of invasive breast carcinoma. Histologic findings include florid tumor emboli that obstruct dermal lymphatics, which results in swelling and inflammation of the affected breast.
Inflammatory breast cancer has been associated with a poor prognosis. However, treatment advances are helping to improve outcomes. Currently, 5-year survival rates are reported to be 40%-70%, with a median survival of 2-4 years. According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the first-line treatment of inflammatory breast cancer involves neoadjuvant chemotherapy, modified radical mastectomy, and adjuvant radiation to the chest wall and regional nodes. Endocrine treatment should also be given to patients who are ER-positive and/or PR-positive (sequential chemotherapy followed by endocrine therapy). For patients who are HER2-positive, up to 1 year of HER2-targeted therapy should be given. HER2-targeted therapies can be administered concurrently with radiation and with endocrine therapy if indicated.
Delayed reconstruction after mastectomy remains the clinical standard for inflammatory breast cancer. This is because the need to resect involved skin negates the benefit of skin-sparing mastectomy for immediate reconstruction. Moreover, high rates of local and distant recurrence warrant comprehensive regional node irradiation in a timely fashion, which may be more challenging or subject to delay after immediate reconstruction. Rarely, the extent of skin excision at the time of mastectomy prohibits primary or local closure. In such cases, reconstruction of the chest wall defect with autologous tissue is required, and concomitant immediate reconstruction may be undertaken.
Detailed guidance on the treatment of inflammatory breast cancer, in the first line and beyond, are available from the NCCN.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of inflammatory breast cancer.
Breast cancer is the leading life-threatening cancer diagnosed and the second-leading cause of cancer-related deaths in women worldwide. In the United States, estimates suggest that 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 women died of the disease. Globally, approximately 2.3 million new diagnoses and 685,000 breast cancer–related deaths were reported in 2020.
Inflammatory breast cancer is a rare and highly aggressive subtype of locally advanced breast cancer. In the United States, inflammatory breast cancer accounts for approximately 2%-4% of breast cancer cases. Although its incidence is rare, 7% of breast cancer caused mortality is attributed to inflammatory breast cancer. Cases of inflammatory breast cancer tend to be diagnosed at a younger age compared with noninflammatory breast cancer cases. Risk factors include African-American race and obesity.
The symptoms of inflammatory breast cancer can vary broadly, ranging from subtle skin erythema to diffuse breast involvement with skin dimpling and nipple inversion. Diagnostic criteria include erythema occupying at least one third of the breast, edema, peau d'orange, and/or warmth, with or without an underlying mass; rapid onset (< 3 months); and pathologic confirmation of invasive breast carcinoma. Histologic findings include florid tumor emboli that obstruct dermal lymphatics, which results in swelling and inflammation of the affected breast.
Inflammatory breast cancer has been associated with a poor prognosis. However, treatment advances are helping to improve outcomes. Currently, 5-year survival rates are reported to be 40%-70%, with a median survival of 2-4 years. According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the first-line treatment of inflammatory breast cancer involves neoadjuvant chemotherapy, modified radical mastectomy, and adjuvant radiation to the chest wall and regional nodes. Endocrine treatment should also be given to patients who are ER-positive and/or PR-positive (sequential chemotherapy followed by endocrine therapy). For patients who are HER2-positive, up to 1 year of HER2-targeted therapy should be given. HER2-targeted therapies can be administered concurrently with radiation and with endocrine therapy if indicated.
Delayed reconstruction after mastectomy remains the clinical standard for inflammatory breast cancer. This is because the need to resect involved skin negates the benefit of skin-sparing mastectomy for immediate reconstruction. Moreover, high rates of local and distant recurrence warrant comprehensive regional node irradiation in a timely fashion, which may be more challenging or subject to delay after immediate reconstruction. Rarely, the extent of skin excision at the time of mastectomy prohibits primary or local closure. In such cases, reconstruction of the chest wall defect with autologous tissue is required, and concomitant immediate reconstruction may be undertaken.
Detailed guidance on the treatment of inflammatory breast cancer, in the first line and beyond, are available from the NCCN.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
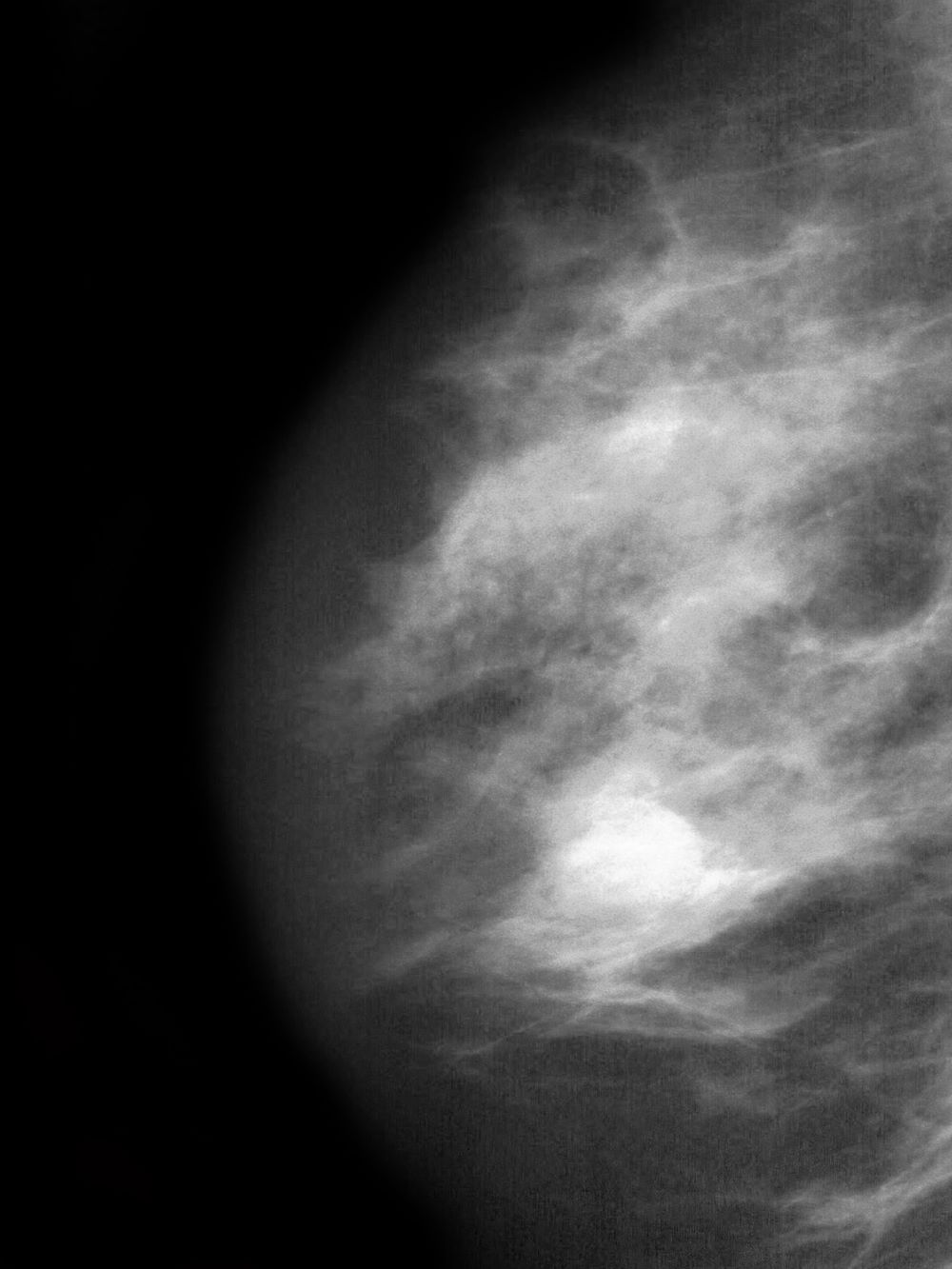
A 51-year-old nonsmoking Black woman presents with a lump in her left breast, as well as associated skin changes and pain of approximately 3 months' duration. The patient last underwent routine screening breast imaging 2 years earlier. The patient is 5 ft 7 in and weighs 200 lb (BMI 31.3). Previous medical history is unremarkable. There is a family history of breast cancer (maternal aunt) and lung cancer (maternal uncle). Physical examination reveals a palpable abnormality in the left breast with edema, skin thickening, and peau d'orange. More than one third of the breast is erythematous. A bilateral mammography reveals an irregular mass and calcifications in the upper outer quadrant of the left breast as well as numerous additional masses and focal asymmetries involving the upper outer and lower outer quadrant of the left breast that extend into the inner left breast. A 1.6-cm mass in the upper left breast is noted, with total abnormality spanning 12.7 cm. Left axillary lymphadenopathy is also observed. Skin punch biopsy of the affected breast reveals dermal lymphatic invasion by tumor cells and tumor emboli. Left axial fine-needle aspiration biopsy reveals malignant cells.
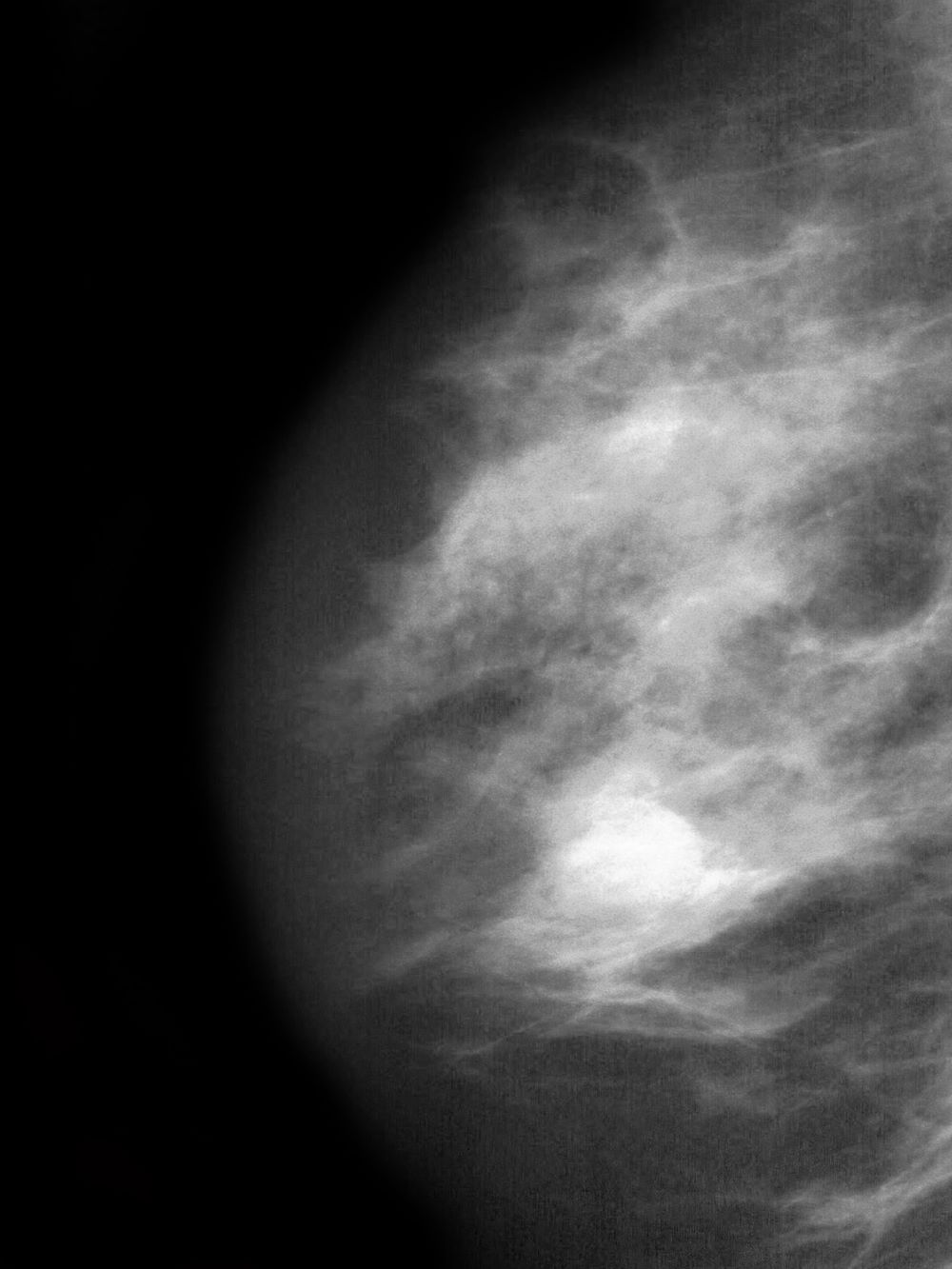
Pruritus and swelling
The history and findings in this case are suggestive of chronic kidney disease (CKD).
CKD affects between 8% and 16% of the population worldwide. Risk factors for CKD are numerous and include T2D, hypertension, and prediabetes. Diabetes is the leading cause of CKD. Up to 40% of patients with diabetes develop diabetic kidney disease, which can progress to end-stage renal disease (ESRD) requiring dialysis or kidney transplantation. In fact, diabetic kidney disease is the top cause of ESRD in the United States.
Diagnostic criteria for CKD include elevated urinary albumin excretion (albuminuria) and/or eGFR < 60 mL/1.73 m2 that persists for more than 3 months. The normal presentation of diabetic kidney disease includes long-standing diabetes, retinopathy, albuminuria without gross hematuria, and gradually progressive decline of eGFR. However, signs of diabetic kidney disease may be present in patients at diagnosis or without retinopathy in T2D. Reduced eGFR without albuminuria has been frequently reported in both type 1 diabetes (T1D) and T2D and is becoming increasingly common as the prevalence of diabetes rises in the United States.
Chronic kidney disease is usually identified through routine screening with serum chemistry profile and urine studies or as an incidental finding. Less often, patients may present with symptoms, such as gross hematuria, "foamy urine" (a sign of albuminuria), nocturia, flank pain, or decreased urine output. In advanced cases, patients may report fatigue, poor appetite, nausea, vomiting, a metallic taste, unintentional weight loss, pruritus, changes in mental status, dyspnea, and/or peripheral edema.
The American Diabetes Association (ADA) 2023 Standards of Care in Diabetes describes five stages of CKD. Stages 1-2 are defined by evidence of high albuminuria with eGFR ≥ 60 mL/min/1.73 m2, while stages 3-5 are defined by progressively lower ranges of eGFR. Of note, at any eGFR, the degree of albuminuria is associated with risk for cardiovascular disease, CKD progression, and mortality. Thus, as noted by the ADA Standards, both eGFR and albuminuria should be used to guide treatment decisions; additionally, eGFR levels are essential for modifying drug dosages or restrictions of use, and the degree of albuminuria should influence selection of antihypertensive agents and glucose-lowering medications.
According to the ADA 2023 Standards of Care in Diabetes, for people with non–dialysis-dependent CKD, dietary protein intake should be ∼0.8 g/kg body weight per day (the recommended daily allowance), as this level has been shown to slow GFR decline compared with higher levels of dietary protein intake, with evidence of a greater effect over time. Conversely, higher levels of dietary protein intake (> 20% of daily calories from protein or > 1.3 g/kg/d) have been associated with increased albuminuria, more rapid kidney function loss, and cardiovascular disease mortality. For patients on dialysis, higher levels of dietary protein intake should be considered, because malnutrition is a significant problem in some of these patients.
Urinary excretion of sodium and potassium may be impaired in patients with reduced eGFR. Thus, restriction of dietary sodium to < 2300 mg/d may help to control blood pressure and reduce cardiovascular risk, and restriction of dietary potassium may be necessary to control serum potassium concentration.
Intensive glycemic control with the goal of achieving near-normoglycemia has been shown to delay the onset and progression of albuminuria and reduced eGFR in patients with diabetes. Insulin alone was used to lower blood glucose in the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study of T1D while a variety of agents were used in clinical trials of T2D, supporting the conclusion that glycemic control itself helps prevent CKD and its progression. However, the presence of CKD affects the risks and benefits of intensive glycemic control and several glucose-lowering medications. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial of T2D, increased adverse effects of intensive glycemic control (hypoglycemia and mortality) were seen among patients with kidney disease at baseline. Moreover, it may take at least 2 years to see improved eGFR outcomes as an effect of intensive glycemic control. Therefore, in some patients with prevalent CKD and substantial comorbidity, target A1c levels may be less intensive.
According to guidance from the US Food and Drug Administration, eGFR should be monitored while taking metformin and metformin is contraindicated in patients with an eGFR < 30 mL/min/1.73 m2. Clinicians should assess the benefits and risks of continuing treatment when eGFR falls to < 45 mL/min/1.73 m2.
The ADA recommends that sodium–glucose cotransporter 2 inhibitors be given to all patients with stage 3 CKD or higher and T2D, regardless of glycemic control, as they have been shown to delay CKD progression and reduce heart failure risk independent of glycemic control. Glucagon-like peptide 1 receptor agonists (GLP-1 RAs) also have direct effects on the kidney and have been reported to improve renal outcomes compared with placebo. In patients for whom cardiovascular risk is a predominant problem, the ADA suggests using GLP-1 RAs for cardiovascular risk reduction.
Comprehensive guidance on the management of CKD in patients with T2D is available in the ADA 2023 Standards of Care in Diabetes.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of chronic kidney disease (CKD).
CKD affects between 8% and 16% of the population worldwide. Risk factors for CKD are numerous and include T2D, hypertension, and prediabetes. Diabetes is the leading cause of CKD. Up to 40% of patients with diabetes develop diabetic kidney disease, which can progress to end-stage renal disease (ESRD) requiring dialysis or kidney transplantation. In fact, diabetic kidney disease is the top cause of ESRD in the United States.
Diagnostic criteria for CKD include elevated urinary albumin excretion (albuminuria) and/or eGFR < 60 mL/1.73 m2 that persists for more than 3 months. The normal presentation of diabetic kidney disease includes long-standing diabetes, retinopathy, albuminuria without gross hematuria, and gradually progressive decline of eGFR. However, signs of diabetic kidney disease may be present in patients at diagnosis or without retinopathy in T2D. Reduced eGFR without albuminuria has been frequently reported in both type 1 diabetes (T1D) and T2D and is becoming increasingly common as the prevalence of diabetes rises in the United States.
Chronic kidney disease is usually identified through routine screening with serum chemistry profile and urine studies or as an incidental finding. Less often, patients may present with symptoms, such as gross hematuria, "foamy urine" (a sign of albuminuria), nocturia, flank pain, or decreased urine output. In advanced cases, patients may report fatigue, poor appetite, nausea, vomiting, a metallic taste, unintentional weight loss, pruritus, changes in mental status, dyspnea, and/or peripheral edema.
The American Diabetes Association (ADA) 2023 Standards of Care in Diabetes describes five stages of CKD. Stages 1-2 are defined by evidence of high albuminuria with eGFR ≥ 60 mL/min/1.73 m2, while stages 3-5 are defined by progressively lower ranges of eGFR. Of note, at any eGFR, the degree of albuminuria is associated with risk for cardiovascular disease, CKD progression, and mortality. Thus, as noted by the ADA Standards, both eGFR and albuminuria should be used to guide treatment decisions; additionally, eGFR levels are essential for modifying drug dosages or restrictions of use, and the degree of albuminuria should influence selection of antihypertensive agents and glucose-lowering medications.
According to the ADA 2023 Standards of Care in Diabetes, for people with non–dialysis-dependent CKD, dietary protein intake should be ∼0.8 g/kg body weight per day (the recommended daily allowance), as this level has been shown to slow GFR decline compared with higher levels of dietary protein intake, with evidence of a greater effect over time. Conversely, higher levels of dietary protein intake (> 20% of daily calories from protein or > 1.3 g/kg/d) have been associated with increased albuminuria, more rapid kidney function loss, and cardiovascular disease mortality. For patients on dialysis, higher levels of dietary protein intake should be considered, because malnutrition is a significant problem in some of these patients.
Urinary excretion of sodium and potassium may be impaired in patients with reduced eGFR. Thus, restriction of dietary sodium to < 2300 mg/d may help to control blood pressure and reduce cardiovascular risk, and restriction of dietary potassium may be necessary to control serum potassium concentration.
Intensive glycemic control with the goal of achieving near-normoglycemia has been shown to delay the onset and progression of albuminuria and reduced eGFR in patients with diabetes. Insulin alone was used to lower blood glucose in the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study of T1D while a variety of agents were used in clinical trials of T2D, supporting the conclusion that glycemic control itself helps prevent CKD and its progression. However, the presence of CKD affects the risks and benefits of intensive glycemic control and several glucose-lowering medications. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial of T2D, increased adverse effects of intensive glycemic control (hypoglycemia and mortality) were seen among patients with kidney disease at baseline. Moreover, it may take at least 2 years to see improved eGFR outcomes as an effect of intensive glycemic control. Therefore, in some patients with prevalent CKD and substantial comorbidity, target A1c levels may be less intensive.
According to guidance from the US Food and Drug Administration, eGFR should be monitored while taking metformin and metformin is contraindicated in patients with an eGFR < 30 mL/min/1.73 m2. Clinicians should assess the benefits and risks of continuing treatment when eGFR falls to < 45 mL/min/1.73 m2.
The ADA recommends that sodium–glucose cotransporter 2 inhibitors be given to all patients with stage 3 CKD or higher and T2D, regardless of glycemic control, as they have been shown to delay CKD progression and reduce heart failure risk independent of glycemic control. Glucagon-like peptide 1 receptor agonists (GLP-1 RAs) also have direct effects on the kidney and have been reported to improve renal outcomes compared with placebo. In patients for whom cardiovascular risk is a predominant problem, the ADA suggests using GLP-1 RAs for cardiovascular risk reduction.
Comprehensive guidance on the management of CKD in patients with T2D is available in the ADA 2023 Standards of Care in Diabetes.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of chronic kidney disease (CKD).
CKD affects between 8% and 16% of the population worldwide. Risk factors for CKD are numerous and include T2D, hypertension, and prediabetes. Diabetes is the leading cause of CKD. Up to 40% of patients with diabetes develop diabetic kidney disease, which can progress to end-stage renal disease (ESRD) requiring dialysis or kidney transplantation. In fact, diabetic kidney disease is the top cause of ESRD in the United States.
Diagnostic criteria for CKD include elevated urinary albumin excretion (albuminuria) and/or eGFR < 60 mL/1.73 m2 that persists for more than 3 months. The normal presentation of diabetic kidney disease includes long-standing diabetes, retinopathy, albuminuria without gross hematuria, and gradually progressive decline of eGFR. However, signs of diabetic kidney disease may be present in patients at diagnosis or without retinopathy in T2D. Reduced eGFR without albuminuria has been frequently reported in both type 1 diabetes (T1D) and T2D and is becoming increasingly common as the prevalence of diabetes rises in the United States.
Chronic kidney disease is usually identified through routine screening with serum chemistry profile and urine studies or as an incidental finding. Less often, patients may present with symptoms, such as gross hematuria, "foamy urine" (a sign of albuminuria), nocturia, flank pain, or decreased urine output. In advanced cases, patients may report fatigue, poor appetite, nausea, vomiting, a metallic taste, unintentional weight loss, pruritus, changes in mental status, dyspnea, and/or peripheral edema.
The American Diabetes Association (ADA) 2023 Standards of Care in Diabetes describes five stages of CKD. Stages 1-2 are defined by evidence of high albuminuria with eGFR ≥ 60 mL/min/1.73 m2, while stages 3-5 are defined by progressively lower ranges of eGFR. Of note, at any eGFR, the degree of albuminuria is associated with risk for cardiovascular disease, CKD progression, and mortality. Thus, as noted by the ADA Standards, both eGFR and albuminuria should be used to guide treatment decisions; additionally, eGFR levels are essential for modifying drug dosages or restrictions of use, and the degree of albuminuria should influence selection of antihypertensive agents and glucose-lowering medications.
According to the ADA 2023 Standards of Care in Diabetes, for people with non–dialysis-dependent CKD, dietary protein intake should be ∼0.8 g/kg body weight per day (the recommended daily allowance), as this level has been shown to slow GFR decline compared with higher levels of dietary protein intake, with evidence of a greater effect over time. Conversely, higher levels of dietary protein intake (> 20% of daily calories from protein or > 1.3 g/kg/d) have been associated with increased albuminuria, more rapid kidney function loss, and cardiovascular disease mortality. For patients on dialysis, higher levels of dietary protein intake should be considered, because malnutrition is a significant problem in some of these patients.
Urinary excretion of sodium and potassium may be impaired in patients with reduced eGFR. Thus, restriction of dietary sodium to < 2300 mg/d may help to control blood pressure and reduce cardiovascular risk, and restriction of dietary potassium may be necessary to control serum potassium concentration.
Intensive glycemic control with the goal of achieving near-normoglycemia has been shown to delay the onset and progression of albuminuria and reduced eGFR in patients with diabetes. Insulin alone was used to lower blood glucose in the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study of T1D while a variety of agents were used in clinical trials of T2D, supporting the conclusion that glycemic control itself helps prevent CKD and its progression. However, the presence of CKD affects the risks and benefits of intensive glycemic control and several glucose-lowering medications. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial of T2D, increased adverse effects of intensive glycemic control (hypoglycemia and mortality) were seen among patients with kidney disease at baseline. Moreover, it may take at least 2 years to see improved eGFR outcomes as an effect of intensive glycemic control. Therefore, in some patients with prevalent CKD and substantial comorbidity, target A1c levels may be less intensive.
According to guidance from the US Food and Drug Administration, eGFR should be monitored while taking metformin and metformin is contraindicated in patients with an eGFR < 30 mL/min/1.73 m2. Clinicians should assess the benefits and risks of continuing treatment when eGFR falls to < 45 mL/min/1.73 m2.
The ADA recommends that sodium–glucose cotransporter 2 inhibitors be given to all patients with stage 3 CKD or higher and T2D, regardless of glycemic control, as they have been shown to delay CKD progression and reduce heart failure risk independent of glycemic control. Glucagon-like peptide 1 receptor agonists (GLP-1 RAs) also have direct effects on the kidney and have been reported to improve renal outcomes compared with placebo. In patients for whom cardiovascular risk is a predominant problem, the ADA suggests using GLP-1 RAs for cardiovascular risk reduction.
Comprehensive guidance on the management of CKD in patients with T2D is available in the ADA 2023 Standards of Care in Diabetes.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
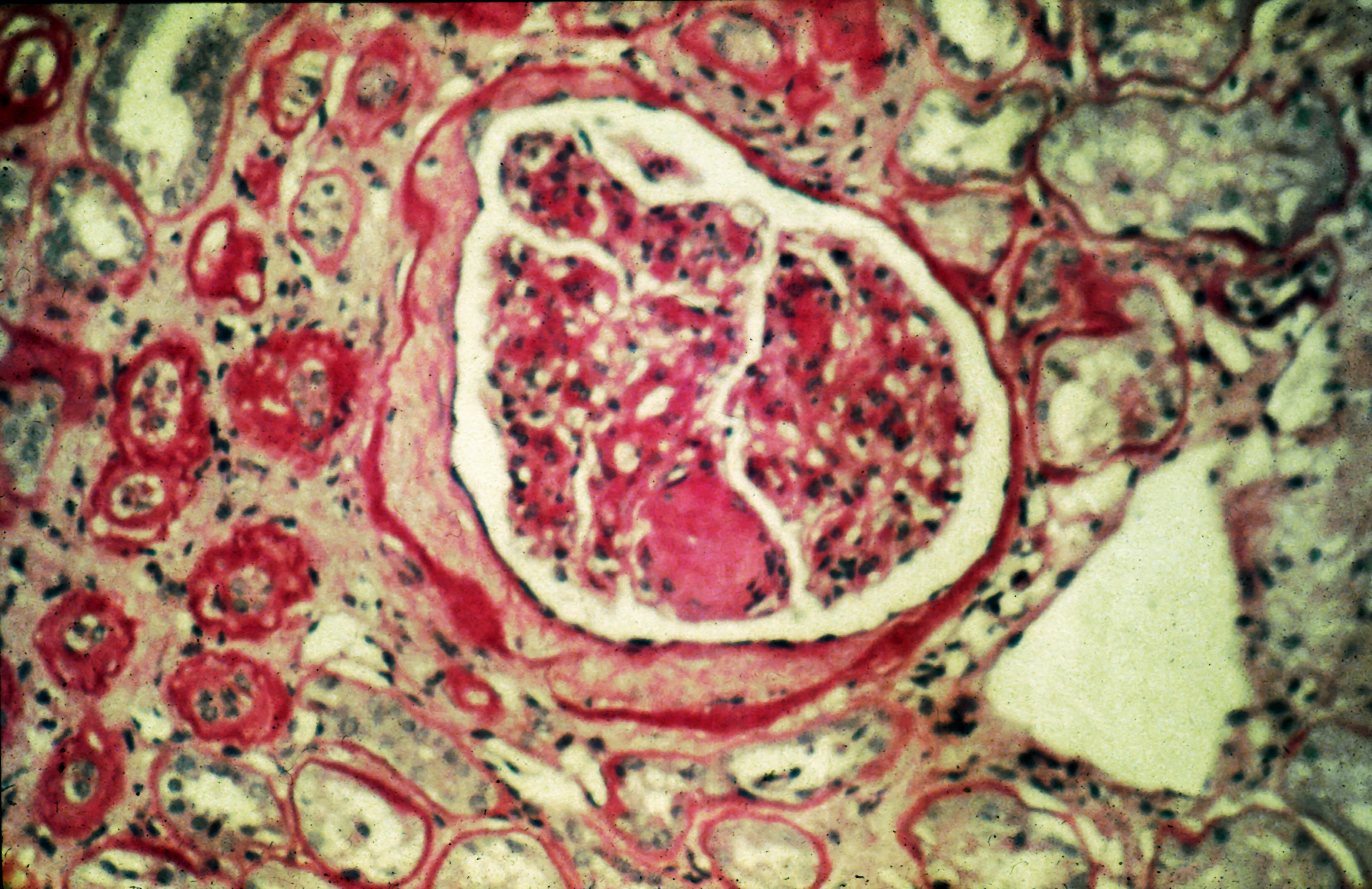
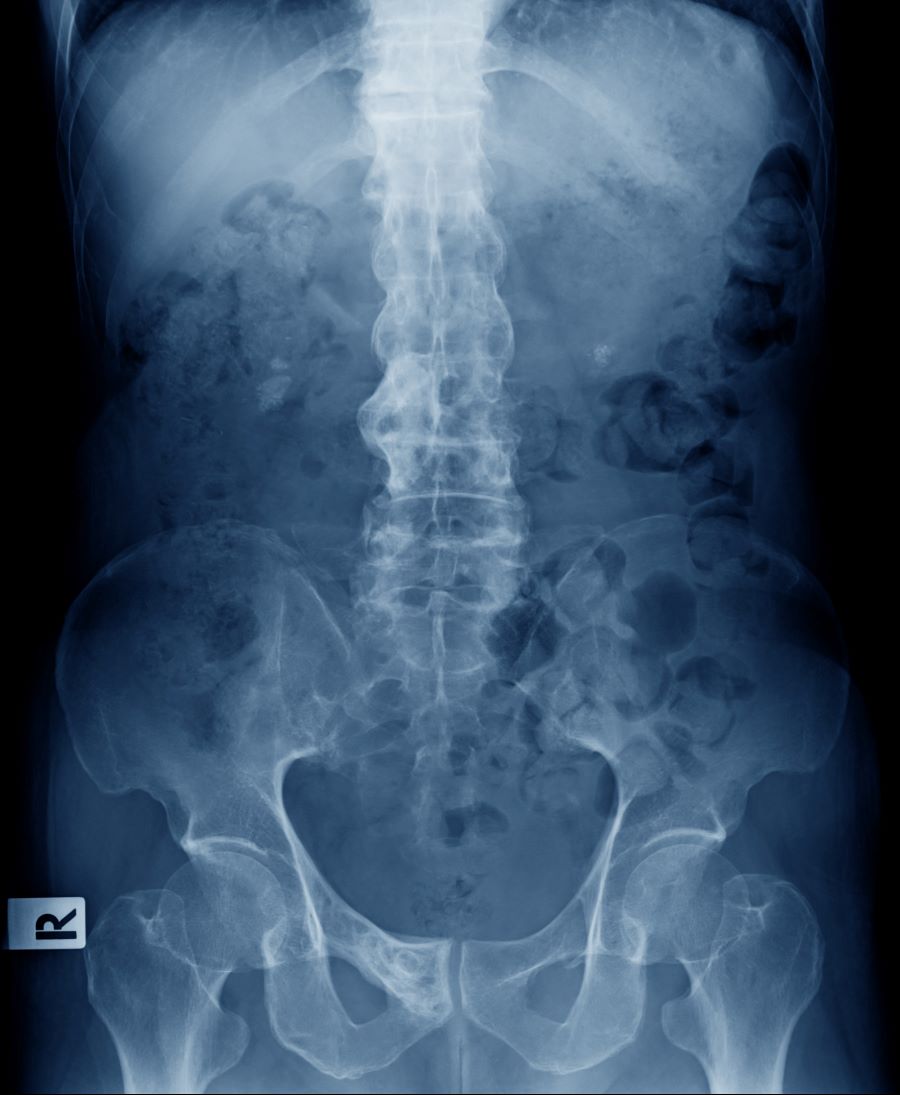
A 56-year-old Hispanic man presents and reports a 2-month history of fatigue, loss of appetite, pruritus, and swelling of the legs, ankles, and feet. The patient was diagnosed with type 2 diabetes (T2D), hypertension, and hyperlipidemia 7 years ago after an ophthalmologist diagnosed him with diabetic retinopathy and referred him for medical care. Since then, he has been inconsistent with attending regular follow-up visits. He is a current smoker (40-pack/year history).
At today's visit, the patient's blood pressure is 150/95 mm Hg, heart rate is 97 beats/min, and respiration rate is 29 breaths/min. He is 5 ft 9 in and weighs 210 lb (BMI 31). Current medications include metformin ER 1000 mg/d, atorvastatin 40 mg/d, amlodipine 10 mg/d, and hydrochlorothiazide 25 mg/d. At a routine visit 4 months ago, the patient's estimated glomerular filtration rate (eGFR) was 59 mL/min/1.73 m2; at a subsequent follow-up visit, his eGFR was 57 mL/min/1.73 m2.
Pertinent laboratory findings today include eGFR 56 mL/min/1.73 m2, serum creatinine 2.7 g/dL, serum albumin 3.3 g/dL, A1c 8.8%, glucose 189 mg/dL, and an albumin-creatinine ratio of 225 mg/g. All other findings are within normal ranges.

Worsening cognitive impairments
The history and findings in this case are suggestive of Alzheimer's disease (AD).
AD is the most common type of dementia. It is characterized by cognitive and behavioral impairment that significantly impairs a patient's social and occupational functioning. The predominant AD pathogenesis hypothesis suggests that AD is largely caused by the accumulation of insoluble amyloid beta deposits and neurofibrillary tangles induced by highly phosphorylated tau proteins in the neocortex, hippocampus, and amygdala, as well as significant loss of neurons and synapses, which leads to brain atrophy. Estimates suggest that approximately 6.2 million people ≥ 65 years of age have AD and that by 2060, the number of Americans with AD may increase to 13.8 million, the result of an aging population and the lack of effective prevention and treatment strategies. AD is a chronic disease that confers tremendous emotional and economic burdens to individuals, families, and society.
Insidiously progressive memory loss is commonly seen in patients presenting with AD. As the disease progresses over the course of several years, other areas of cognition are impaired. Patients may develop language disorders (eg, anomic aphasia or anomia) and impairment in visuospatial skills and executive functions. Slowly progressive behavioral changes are also observed in many individuals with AD.
Criteria for the clinical diagnosis of AD (eg, insidious onset of cognitive impairment, clear history of worsening symptoms) have been developed and are frequently employed. Among individuals who meet the core clinical criteria for probable AD dementia, biomarker evidence may help to increase the certainty that AD is the basis of the clinical dementia syndrome. Several cerebrospinal fluid and blood biomarkers have shown excellent diagnostic ability by identifying tau pathology and cerebral amyloid beta for AD. Neuroimaging is becoming increasingly important for identifying the underlying causes of cognitive impairment. Currently, MRI is considered the preferred neuroimaging modality for AD as it enables accurate measurement of the three-dimensional volume of brain structures, particularly the size of the hippocampus and related regions. CT may be used when MRI is not possible, such as in a patient with a pacemaker.
PET is increasingly being used as a noninvasive method for depicting tau pathology deposition and distribution in patients with cognitive impairment. In 2020, the US Food and Drug Administration approved the first tau PET tracer, 18F-flortaucipir, a significant achievement in improving AD diagnosis.
Currently, the only therapies available for AD are symptomatic therapies. Cholinesterase inhibitors and a partial N-methyl-d-aspartate antagonist are the standard medical treatment for AD. Recently approved antiamyloid therapies are also available for patients with mild cognitive impairment or mild dementia. These include aducanumab, a first-in-class amyloid beta–directed antibody that was approved in 2021; and lecanemab, another amyloid beta–directed antibody that was approved in 2023. Both aducanumab and lecanemab are recommended for the treatment of patients with mild cognitive impairment or mild dementia stage of disease, the population in which the safety and efficacy of these newer agents were demonstrated in clinical trials.
Psychotropic agents are often used to treat the secondary symptoms of AD, such as depression, agitation, aggression, hallucinations, delusions, and/or sleep disorders, which can be problematic. Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of Alzheimer's disease (AD).
AD is the most common type of dementia. It is characterized by cognitive and behavioral impairment that significantly impairs a patient's social and occupational functioning. The predominant AD pathogenesis hypothesis suggests that AD is largely caused by the accumulation of insoluble amyloid beta deposits and neurofibrillary tangles induced by highly phosphorylated tau proteins in the neocortex, hippocampus, and amygdala, as well as significant loss of neurons and synapses, which leads to brain atrophy. Estimates suggest that approximately 6.2 million people ≥ 65 years of age have AD and that by 2060, the number of Americans with AD may increase to 13.8 million, the result of an aging population and the lack of effective prevention and treatment strategies. AD is a chronic disease that confers tremendous emotional and economic burdens to individuals, families, and society.
Insidiously progressive memory loss is commonly seen in patients presenting with AD. As the disease progresses over the course of several years, other areas of cognition are impaired. Patients may develop language disorders (eg, anomic aphasia or anomia) and impairment in visuospatial skills and executive functions. Slowly progressive behavioral changes are also observed in many individuals with AD.
Criteria for the clinical diagnosis of AD (eg, insidious onset of cognitive impairment, clear history of worsening symptoms) have been developed and are frequently employed. Among individuals who meet the core clinical criteria for probable AD dementia, biomarker evidence may help to increase the certainty that AD is the basis of the clinical dementia syndrome. Several cerebrospinal fluid and blood biomarkers have shown excellent diagnostic ability by identifying tau pathology and cerebral amyloid beta for AD. Neuroimaging is becoming increasingly important for identifying the underlying causes of cognitive impairment. Currently, MRI is considered the preferred neuroimaging modality for AD as it enables accurate measurement of the three-dimensional volume of brain structures, particularly the size of the hippocampus and related regions. CT may be used when MRI is not possible, such as in a patient with a pacemaker.
PET is increasingly being used as a noninvasive method for depicting tau pathology deposition and distribution in patients with cognitive impairment. In 2020, the US Food and Drug Administration approved the first tau PET tracer, 18F-flortaucipir, a significant achievement in improving AD diagnosis.
Currently, the only therapies available for AD are symptomatic therapies. Cholinesterase inhibitors and a partial N-methyl-d-aspartate antagonist are the standard medical treatment for AD. Recently approved antiamyloid therapies are also available for patients with mild cognitive impairment or mild dementia. These include aducanumab, a first-in-class amyloid beta–directed antibody that was approved in 2021; and lecanemab, another amyloid beta–directed antibody that was approved in 2023. Both aducanumab and lecanemab are recommended for the treatment of patients with mild cognitive impairment or mild dementia stage of disease, the population in which the safety and efficacy of these newer agents were demonstrated in clinical trials.
Psychotropic agents are often used to treat the secondary symptoms of AD, such as depression, agitation, aggression, hallucinations, delusions, and/or sleep disorders, which can be problematic. Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of Alzheimer's disease (AD).
AD is the most common type of dementia. It is characterized by cognitive and behavioral impairment that significantly impairs a patient's social and occupational functioning. The predominant AD pathogenesis hypothesis suggests that AD is largely caused by the accumulation of insoluble amyloid beta deposits and neurofibrillary tangles induced by highly phosphorylated tau proteins in the neocortex, hippocampus, and amygdala, as well as significant loss of neurons and synapses, which leads to brain atrophy. Estimates suggest that approximately 6.2 million people ≥ 65 years of age have AD and that by 2060, the number of Americans with AD may increase to 13.8 million, the result of an aging population and the lack of effective prevention and treatment strategies. AD is a chronic disease that confers tremendous emotional and economic burdens to individuals, families, and society.
Insidiously progressive memory loss is commonly seen in patients presenting with AD. As the disease progresses over the course of several years, other areas of cognition are impaired. Patients may develop language disorders (eg, anomic aphasia or anomia) and impairment in visuospatial skills and executive functions. Slowly progressive behavioral changes are also observed in many individuals with AD.
Criteria for the clinical diagnosis of AD (eg, insidious onset of cognitive impairment, clear history of worsening symptoms) have been developed and are frequently employed. Among individuals who meet the core clinical criteria for probable AD dementia, biomarker evidence may help to increase the certainty that AD is the basis of the clinical dementia syndrome. Several cerebrospinal fluid and blood biomarkers have shown excellent diagnostic ability by identifying tau pathology and cerebral amyloid beta for AD. Neuroimaging is becoming increasingly important for identifying the underlying causes of cognitive impairment. Currently, MRI is considered the preferred neuroimaging modality for AD as it enables accurate measurement of the three-dimensional volume of brain structures, particularly the size of the hippocampus and related regions. CT may be used when MRI is not possible, such as in a patient with a pacemaker.
PET is increasingly being used as a noninvasive method for depicting tau pathology deposition and distribution in patients with cognitive impairment. In 2020, the US Food and Drug Administration approved the first tau PET tracer, 18F-flortaucipir, a significant achievement in improving AD diagnosis.
Currently, the only therapies available for AD are symptomatic therapies. Cholinesterase inhibitors and a partial N-methyl-d-aspartate antagonist are the standard medical treatment for AD. Recently approved antiamyloid therapies are also available for patients with mild cognitive impairment or mild dementia. These include aducanumab, a first-in-class amyloid beta–directed antibody that was approved in 2021; and lecanemab, another amyloid beta–directed antibody that was approved in 2023. Both aducanumab and lecanemab are recommended for the treatment of patients with mild cognitive impairment or mild dementia stage of disease, the population in which the safety and efficacy of these newer agents were demonstrated in clinical trials.
Psychotropic agents are often used to treat the secondary symptoms of AD, such as depression, agitation, aggression, hallucinations, delusions, and/or sleep disorders, which can be problematic. Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Behavioral interventions, including patient-centered approaches and caregiver training, may also be beneficial for managing the cognitive and behavioral manifestations of AD. These modalities are often used in combination with pharmacologic interventions, such as anxiolytics for anxiety and agitation, neuroleptics for delusions or hallucinations, and antidepressants or mood stabilizers for mood disorders and specific manifestations (eg, episodes of anger or rage). Regular physical activity and exercise is also emerging as a potential strategy for delaying AD progression and possibly conferring a protective effect on brain health.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
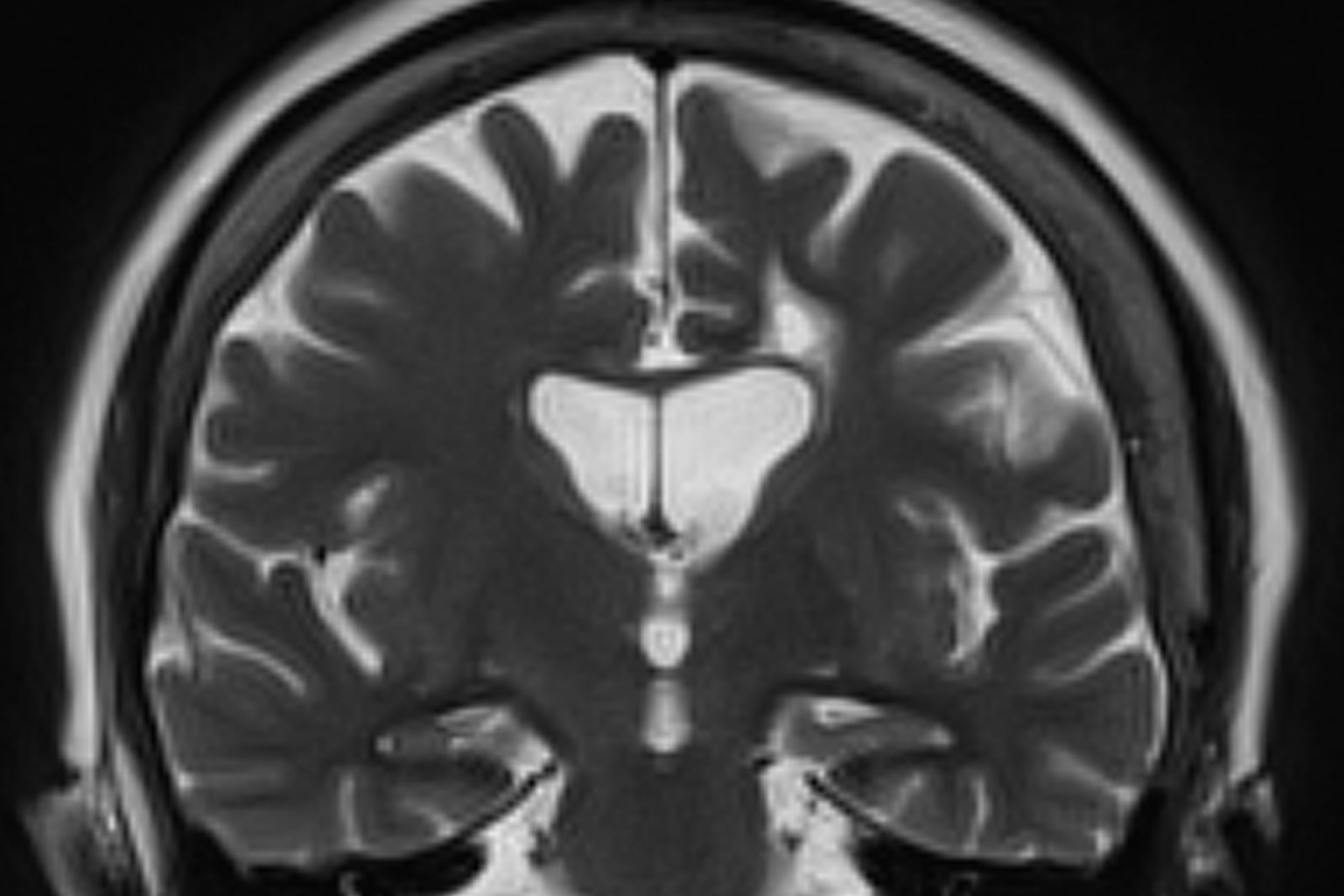
A 73-year-old male restaurant manager presents with concerns of progressively worsening cognitive impairment. The patient's symptoms began approximately 2 years ago. At that time, he attributed them to normal aging. Recently, however, he has begun to have increasing difficulties at work. On several occasions, he has forgotten to place important supply orders and has made errors with staff scheduling. His wife reports that he frequently misplaces items at home, such as his cell phone and car keys, and has been experiencing noticeable deficits with his short-term memory. In addition, he has been "unlike himself" for quite some time, with uncharacteristic episodes of depression, anxiety, and emotional lability. The patient's past medical history is significant for mild obesity, hypertension, and dyslipidemia. There is no history of neurotoxic exposure, head injuries, strokes, or seizures. His family history is negative for dementia. Current medications include rosuvastatin 40 mg/d and metoprolol 100 mg/d. His current height and weight are 5 ft 11 in and 223 lb (BMI 31.1).
No abnormalities are noted on physical exam; the patient's blood pressure, pulse oximetry, and heart rate are within normal ranges. Laboratory tests are within normal ranges, except for elevated levels of fasting blood glucose level (119 mg/dL) and A1c (6.3%). The patient scores 19 on the Montreal Cognitive Assessment test. His clinician orders MRI scanning, which reveals generalized atrophy of brain tissue and an accentuated loss of tissue involving the temporal lobes.
Moderate to severe back pain
The history and findings in this case are suggestive of axial psoriatic arthritis (PsA).
Psoriasis is a complex, chronic, inflammatory, immune-mediated disease that is associated with significant morbidity, reduced quality of life, and increased mortality. Approximately 7.4 million adults in the United States have psoriasis; worldwide, approximately 2%-3% of the population is affected. Patients with psoriasis frequently have comorbidities; PsA, an inflammatory, seronegative musculoskeletal disease, is among the most common. It is estimated that 25%-30% of patients with psoriasis develop PsA.
PsA is a heterogeneous disease. Patients may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Men and women are equally affected by PsA, which typically develops when patients are age 30-50 years. Like psoriasis, PsA is associated with numerous comorbidities, including cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
PsA is a potentially erosive disease. Structural damage and functional impairment occurs within 2 years of initial assessment in approximately 50% of patients; as the disease progresses, patients may experience irreversible joint damage and disability. Axial involvement occurs in 25%-70% of patients with PsA; exclusive axial involvement is uncommon, occurring in 5% of patients. Common symptoms of axial PsA include inflammatory back pain (eg, pain that improves with activity but worsens with rest, morning stiffness lasting longer than 30 minutes). Some patients with axial involvement may be asymptomatic. If untreated, cervical spinal mobility and lateral flexion significantly decline within 5 years in patients with axial PsA. In addition, sacroiliitis worsens over time; 37% and 52% of patients develop grade 2 or higher sacroiliitis within 5 and 10 years, respectively. This highlights the importance of early identification and treatment of patients with axial PsA.
The diagnosis of axial PsA is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosis). PsA can be differentiated from ankylosing spondylitis by the asymmetric and frequently unilateral presentation of sacroiliitis and syndesmophytes, which frequently presents as nonmarginal, bulky, asymmetric, and discontinuous skipping vertebral levels.
Plain radiography, CT, ultrasound, and MRI are all useful tools for evaluating patients with PsA. MRI and ultrasound may be more sensitive than plain radiography is for detecting early joint inflammation and damage as well as axial changes, including sacroiliitis; however, they are not required for a diagnosis of PsA.
The treatment of axial PsA is based on international guidelines developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Assessment of SpondyloArthritis International Society–European League Against Rheumatism. Treatment focuses on minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; enhancing functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Medications for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; however, long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or if erosive disease or other indications of high disease activity is observed, guidelines recommend initiation of a TNF inhibitor. Disease-modifying antirheumatic drugs, such as methotrexate, are not routinely prescribed for patients with axial disease because they have not been shown to be effective.
If symptoms of axial PsA are not controlled by NSAIDs, tumor necrosis factor (TNF) inhibitors are recommended. However, interleukin 17A inhibitors may be used in preference to TNF inhibitors in patients with significant skin involvement. In the United States, adalimumab, certolizumab pegol, golimumab, and infliximab are recommended over etanercept for patients with axial SpA in the presence of concomitant inflammatory bowel disease (IBD) or recurrent uveitis (although there is no evidence for golimumab) because etanercept has contradictory results for uveitis and has not been shown to have efficacy in IBD.
If patients fail to respond to a first trial of a TNF inhibitor, trying a second TNF inhibitor before switching to a different class of biologic is recommended by US guidelines. A Janus kinase inhibitor (tofacitinib) may be considered for patients who do not respond to TNF inhibitors.
Nonpharmacologic therapies (ie, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of axial psoriatic arthritis (PsA).
Psoriasis is a complex, chronic, inflammatory, immune-mediated disease that is associated with significant morbidity, reduced quality of life, and increased mortality. Approximately 7.4 million adults in the United States have psoriasis; worldwide, approximately 2%-3% of the population is affected. Patients with psoriasis frequently have comorbidities; PsA, an inflammatory, seronegative musculoskeletal disease, is among the most common. It is estimated that 25%-30% of patients with psoriasis develop PsA.
PsA is a heterogeneous disease. Patients may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Men and women are equally affected by PsA, which typically develops when patients are age 30-50 years. Like psoriasis, PsA is associated with numerous comorbidities, including cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
PsA is a potentially erosive disease. Structural damage and functional impairment occurs within 2 years of initial assessment in approximately 50% of patients; as the disease progresses, patients may experience irreversible joint damage and disability. Axial involvement occurs in 25%-70% of patients with PsA; exclusive axial involvement is uncommon, occurring in 5% of patients. Common symptoms of axial PsA include inflammatory back pain (eg, pain that improves with activity but worsens with rest, morning stiffness lasting longer than 30 minutes). Some patients with axial involvement may be asymptomatic. If untreated, cervical spinal mobility and lateral flexion significantly decline within 5 years in patients with axial PsA. In addition, sacroiliitis worsens over time; 37% and 52% of patients develop grade 2 or higher sacroiliitis within 5 and 10 years, respectively. This highlights the importance of early identification and treatment of patients with axial PsA.
The diagnosis of axial PsA is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosis). PsA can be differentiated from ankylosing spondylitis by the asymmetric and frequently unilateral presentation of sacroiliitis and syndesmophytes, which frequently presents as nonmarginal, bulky, asymmetric, and discontinuous skipping vertebral levels.
Plain radiography, CT, ultrasound, and MRI are all useful tools for evaluating patients with PsA. MRI and ultrasound may be more sensitive than plain radiography is for detecting early joint inflammation and damage as well as axial changes, including sacroiliitis; however, they are not required for a diagnosis of PsA.
The treatment of axial PsA is based on international guidelines developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Assessment of SpondyloArthritis International Society–European League Against Rheumatism. Treatment focuses on minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; enhancing functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Medications for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; however, long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or if erosive disease or other indications of high disease activity is observed, guidelines recommend initiation of a TNF inhibitor. Disease-modifying antirheumatic drugs, such as methotrexate, are not routinely prescribed for patients with axial disease because they have not been shown to be effective.
If symptoms of axial PsA are not controlled by NSAIDs, tumor necrosis factor (TNF) inhibitors are recommended. However, interleukin 17A inhibitors may be used in preference to TNF inhibitors in patients with significant skin involvement. In the United States, adalimumab, certolizumab pegol, golimumab, and infliximab are recommended over etanercept for patients with axial SpA in the presence of concomitant inflammatory bowel disease (IBD) or recurrent uveitis (although there is no evidence for golimumab) because etanercept has contradictory results for uveitis and has not been shown to have efficacy in IBD.
If patients fail to respond to a first trial of a TNF inhibitor, trying a second TNF inhibitor before switching to a different class of biologic is recommended by US guidelines. A Janus kinase inhibitor (tofacitinib) may be considered for patients who do not respond to TNF inhibitors.
Nonpharmacologic therapies (ie, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of axial psoriatic arthritis (PsA).
Psoriasis is a complex, chronic, inflammatory, immune-mediated disease that is associated with significant morbidity, reduced quality of life, and increased mortality. Approximately 7.4 million adults in the United States have psoriasis; worldwide, approximately 2%-3% of the population is affected. Patients with psoriasis frequently have comorbidities; PsA, an inflammatory, seronegative musculoskeletal disease, is among the most common. It is estimated that 25%-30% of patients with psoriasis develop PsA.
PsA is a heterogeneous disease. Patients may present with nail and skin changes, peripheral arthritis, enthesitis, dactylitis, and axial spondyloarthritis (SpA), either alone or in combination. Men and women are equally affected by PsA, which typically develops when patients are age 30-50 years. Like psoriasis, PsA is associated with numerous comorbidities, including cardiovascular disease, metabolic syndrome, obesity, diabetes, depression, uveitis, and anxiety.
PsA is a potentially erosive disease. Structural damage and functional impairment occurs within 2 years of initial assessment in approximately 50% of patients; as the disease progresses, patients may experience irreversible joint damage and disability. Axial involvement occurs in 25%-70% of patients with PsA; exclusive axial involvement is uncommon, occurring in 5% of patients. Common symptoms of axial PsA include inflammatory back pain (eg, pain that improves with activity but worsens with rest, morning stiffness lasting longer than 30 minutes). Some patients with axial involvement may be asymptomatic. If untreated, cervical spinal mobility and lateral flexion significantly decline within 5 years in patients with axial PsA. In addition, sacroiliitis worsens over time; 37% and 52% of patients develop grade 2 or higher sacroiliitis within 5 and 10 years, respectively. This highlights the importance of early identification and treatment of patients with axial PsA.
The diagnosis of axial PsA is confirmed by physical examination and imaging. Axial PsA characteristics, including sacroiliitis and spondylitis, are distinguished by the development of syndesmophytes (ie, ossification of the annulus fibrosis). PsA can be differentiated from ankylosing spondylitis by the asymmetric and frequently unilateral presentation of sacroiliitis and syndesmophytes, which frequently presents as nonmarginal, bulky, asymmetric, and discontinuous skipping vertebral levels.
Plain radiography, CT, ultrasound, and MRI are all useful tools for evaluating patients with PsA. MRI and ultrasound may be more sensitive than plain radiography is for detecting early joint inflammation and damage as well as axial changes, including sacroiliitis; however, they are not required for a diagnosis of PsA.
The treatment of axial PsA is based on international guidelines developed by the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Assessment of SpondyloArthritis International Society–European League Against Rheumatism. Treatment focuses on minimizing pain, stiffness, and fatigue; improving and preserving spinal flexibility and posture; enhancing functional capacity; and maintaining the ability to work, with a target of remission or minimal/low disease activity.
Medications for symptomatic relief include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and sacroiliac joint injections with glucocorticoids for mild disease; however, long-term treatment with systemic glucocorticoids is not recommended. If patients remain symptomatic or if erosive disease or other indications of high disease activity is observed, guidelines recommend initiation of a TNF inhibitor. Disease-modifying antirheumatic drugs, such as methotrexate, are not routinely prescribed for patients with axial disease because they have not been shown to be effective.
If symptoms of axial PsA are not controlled by NSAIDs, tumor necrosis factor (TNF) inhibitors are recommended. However, interleukin 17A inhibitors may be used in preference to TNF inhibitors in patients with significant skin involvement. In the United States, adalimumab, certolizumab pegol, golimumab, and infliximab are recommended over etanercept for patients with axial SpA in the presence of concomitant inflammatory bowel disease (IBD) or recurrent uveitis (although there is no evidence for golimumab) because etanercept has contradictory results for uveitis and has not been shown to have efficacy in IBD.
If patients fail to respond to a first trial of a TNF inhibitor, trying a second TNF inhibitor before switching to a different class of biologic is recommended by US guidelines. A Janus kinase inhibitor (tofacitinib) may be considered for patients who do not respond to TNF inhibitors.
Nonpharmacologic therapies (ie, exercise, physical therapy, massage therapy, occupational therapy, acupuncture) are recommended for all patients with active PsA.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
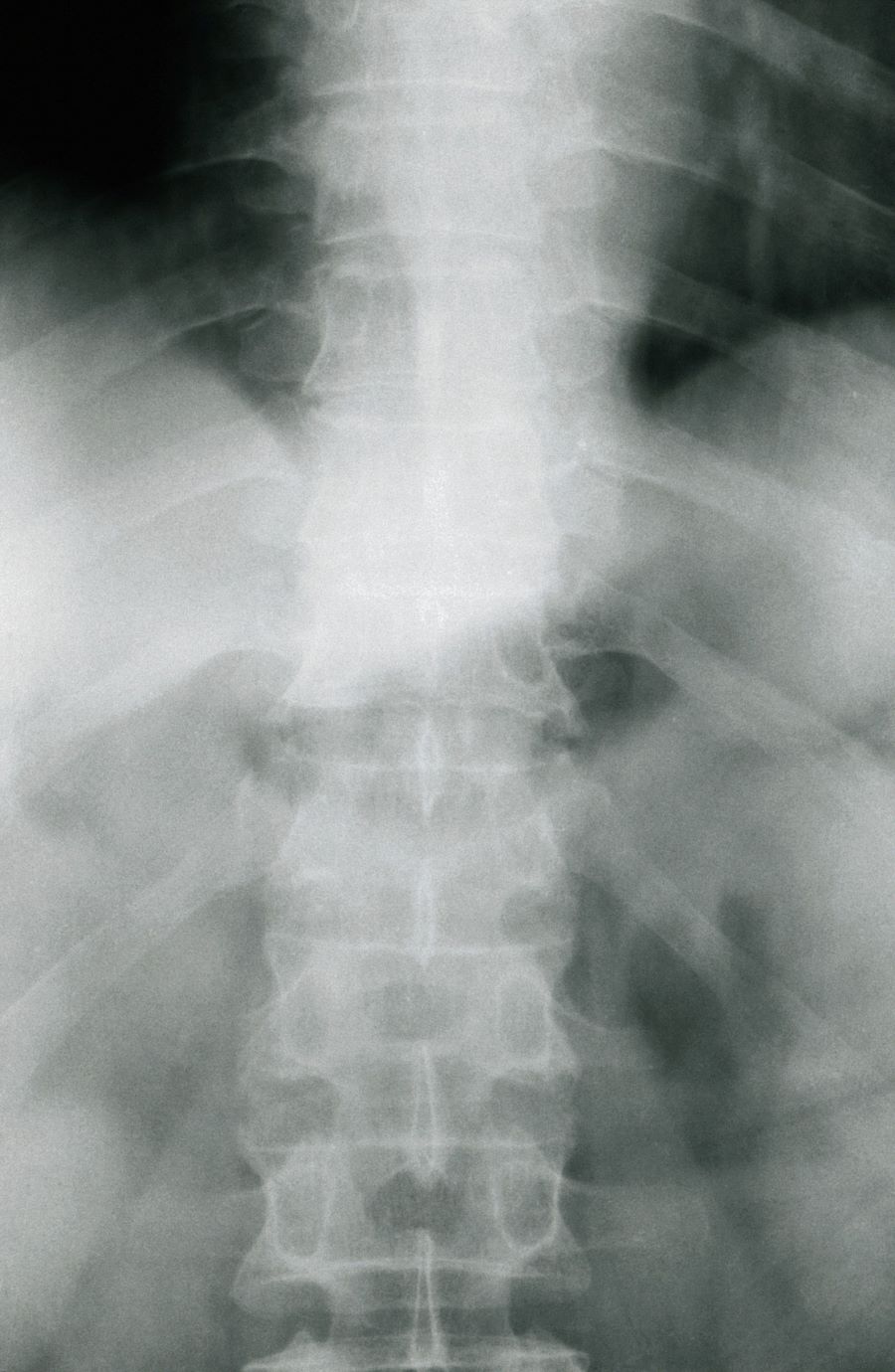
A 38-year-old nonsmoking woman presents with complaints of moderate to severe back pain of approximately 6 months' duration. She also reports morning back/neck stiffness that lasts for approximately 45 minutes and pain/stiffness in her wrists and fingers. The patient states that her back pain improves with exercise (walking and stretching) and worsens in the evening and during long periods of rest. On occasion, she is awakened during the early morning hours because of her back pain. The patient has a 15-year history of moderate to severe psoriasis and a history of irritable bowel disease (IBD). Current medications include cyclosporine 3 mg/d, topical roflumilast 0.3%/d, and loperamide 3 mg as needed. The patient is 5 ft 5 in and weighs 183 lb (BMI of 30.4).
Physical examination reveals psoriatic plaques on the hands, elbows, and knees and nail dystrophy (onycholysis and pitting). Vital signs are within normal ranges. Pertinent laboratory findings include white blood count of 12,000 mcL (> 50% polymorphonuclear leukocytes), erythrocyte sedimentation rate of 19 mm/h, and c-reactive protein of 3 mg/dL. Rheumatoid factor, antinuclear antibody, and anti-citrullinated protein antibody antibody were negative.
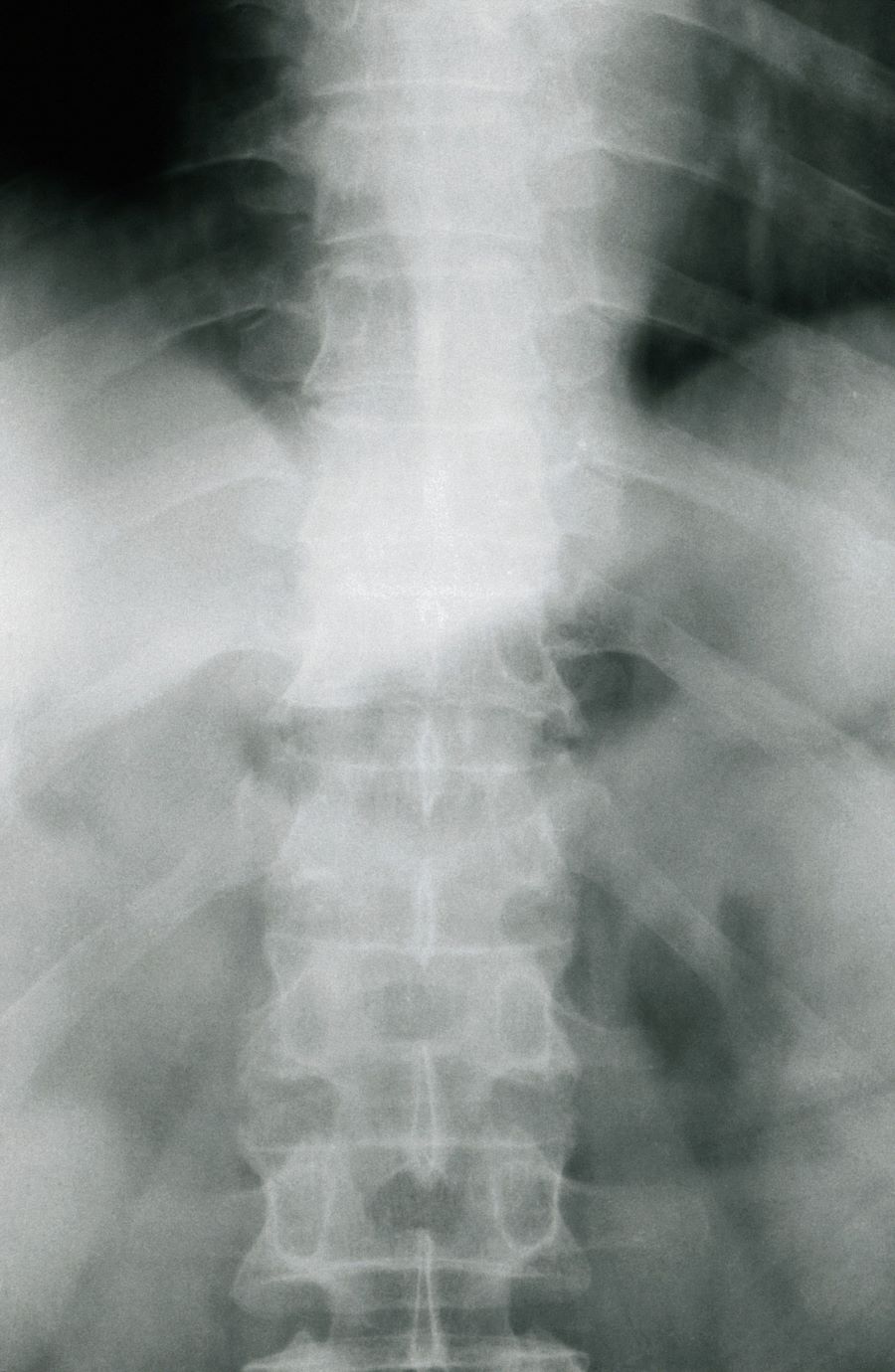
Weight gain and excessive fatigue
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
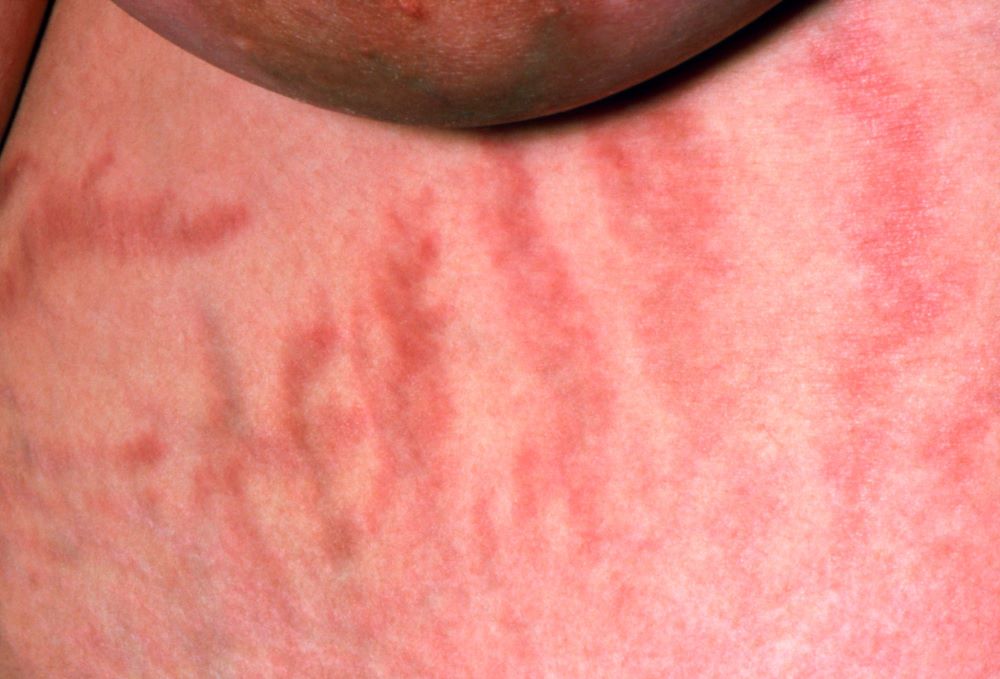
A 37-year-old woman presents with reports of insomnia, weight gain (approximately 12 lb over the last 9 months), and excessive fatigue. The patient's medical history is significant for hypertension (diagnosed 4 years earlier) and depression (diagnosed 7 years earlier). Her current medications include lisinopril 10 mg/d, bupropion 75 mg/d, and venlafaxine 75 mg/d. There is no history of alcohol or drug abuse; family history is unremarkable. The patient's height and weight are 5 ft 5 in and 182 lb (body mass index of 30.3).
During physical examination, facial hirsutism is observed along with increased adipose tissue in the face (moon-shaped face), upper back at the base of the neck (buffalo hump), and abdomen. Vertical red abdominal striae are present. Several bruises are observed on the patient's thighs and arms; when questioned, she reports noting an increased tendency to bruise in recent months.
Pertinent laboratory findings include urinary free cortisol excretion (UFC) 324 mcg/24 h, 1-mg dexamethasone suppression test (DST) with a cortisol value of 3.64 mcg/dL (100.42 nmol/L), and adrenocorticotropic hormone (ACTH) level of 84.9 pg/mL.
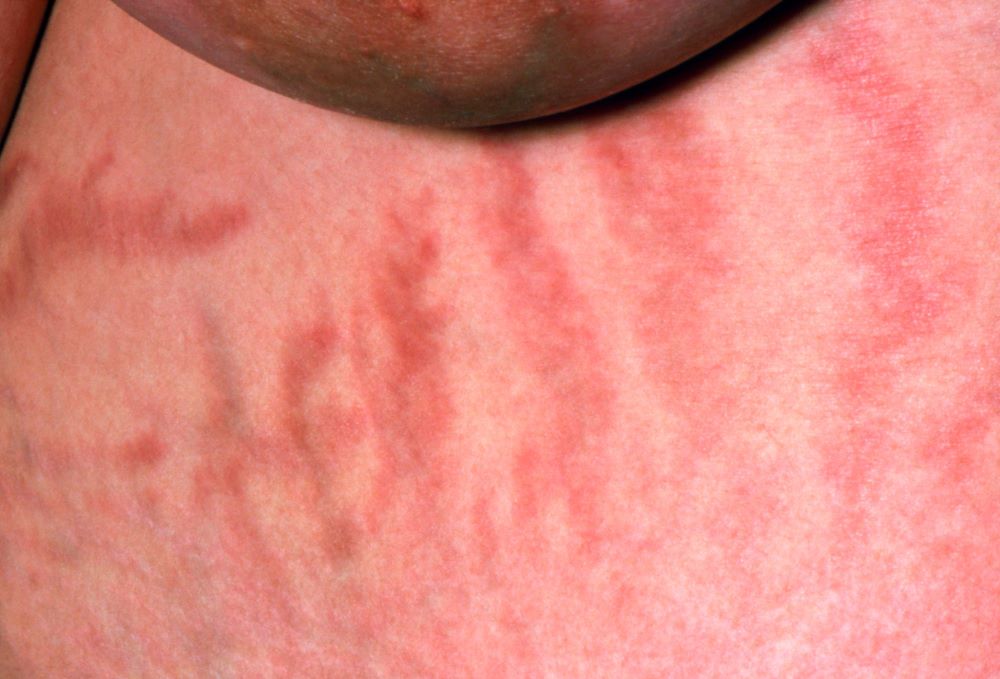
Three-month history of fever
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
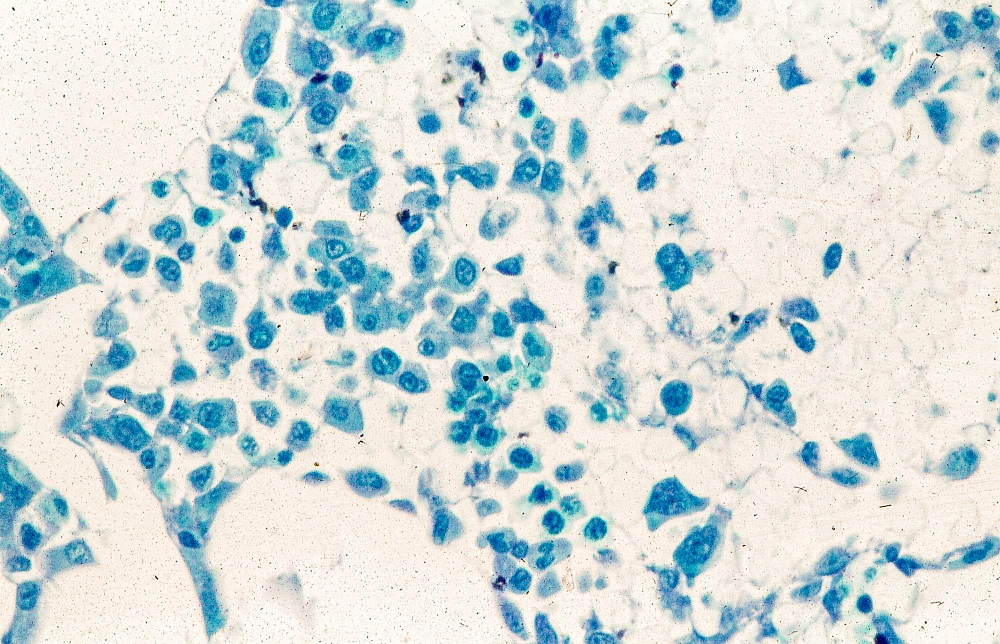
A 62-year-old man with no significant past medical history presents with a 3-month history of fever, night sweats, upper abdominal pain and bloating, and unintentional weight loss. He does not currently take any medications. His height and weight are 6 ft 2 in and 171 lb (BMI 22).
Physical examination reveals generalized lymphadenopathy and splenomegaly. Subsequently, an excisional lymph node biopsy is performed. Histologic examination of the specimen reveals sheets of mostly large cells of varying sizes, with nuclear overlap and extensive necrosis. Cytology findings include large lymphocytes with pale cytoplasm, clumped chromatin, oval irregular nuclei, and prominent nucleoli. Pertinent findings from immunohistochemical staining include the presence of t(11:14), Ki67 > 30%, CD5 and CD20 positivity, and CD10 and CD23 negativity. Centroblasts are absent.
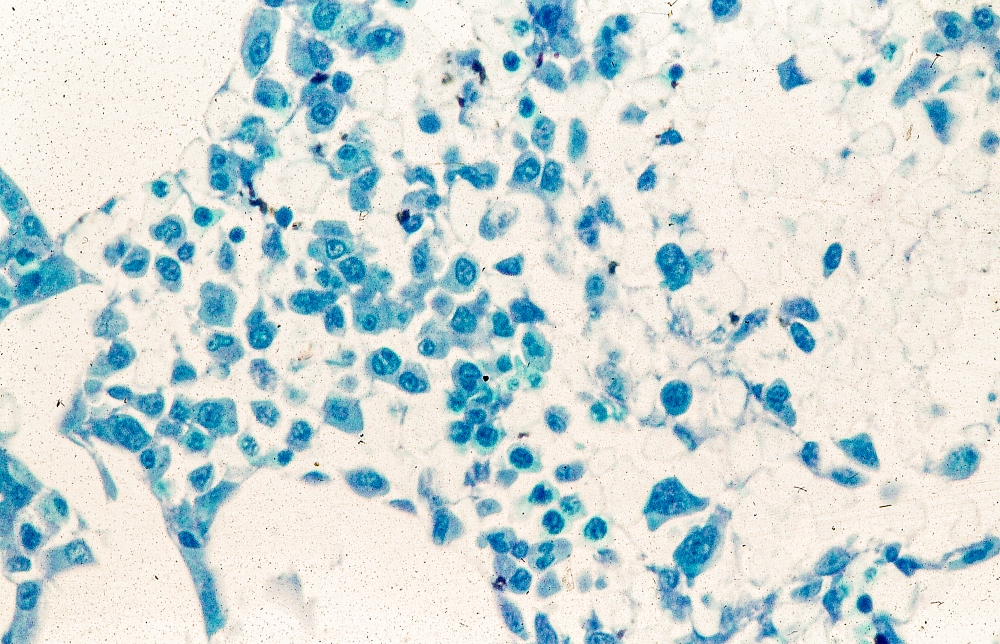
Complaints of cough and fatigue
The history and findings in this case are suggestive of non–small cell lung cancer (NSCLC) large cell carcinoma.
Lung cancer is the most common cancer worldwide and has the highest mortality rate of all cancers. It comprises two major subtypes: NSCLC and small cell lung cancer (SCLC). Histologically, NSCLC is further classified as adenocarcinoma, squamous cell carcinoma, and large cell carcinoma with or without neuroendocrine features. Large cell carcinoma accounts for 9% of all cases and is frequently associated with poor prognosis. Most patients with NSCLC large cell carcinoma are older than 60 years and are diagnosed with stage III or IV disease. NSCLC large cell carcinoma appears to occur more commonly in men than in women and in patients with a history of smoking. It often presents as a large mass with central necrosis.
NSCLC is often asymptomatic in its early stages. The most frequently reported signs and symptoms of lung cancer include:
• Cough
• Chest pain
• Shortness of breath
• Coughing up blood
• Wheezing
• Hoarseness
• Recurring infections, such as bronchitis and pneumonia
• Weight loss and loss of appetite
• Fatigue
Signs and symptoms of metastatic disease may include bone pain, spinal cord impingement, or neurologic problems, such as headache, weakness or numbness of limbs, dizziness, and seizures.
All patients with NSCLC require a complete staging workup to evaluate the extent of disease because stage plays a central role in treatment selection. After physical examination and a complete blood count, a chest radiograph is often the first test performed. Chest radiographs may show a pulmonary nodule, mass, or infiltrate; mediastinal widening; atelectasis; hilar enlargement; and/or pleural effusion.
Various methods are available to confirm the diagnosis, and the method chosen may be determined at least in part by lesion location. These include:
• Bronchoscopy
• Sputum cytology
• Mediastinoscopy
• Thoracentesis
• Thoracoscopy
• Transthoracic needle biopsy (CT- or fluoroscopy-guided)
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the diagnosis of NSCLC large cell carcinoma requires a thoroughly sampled resected tumor with immunohistochemical stains that exclude adenocarcinoma (TTF-1, napsin A) and squamous cell (p40, p63) carcinoma. Nonresected specimens or cytology specimens are insufficient for its diagnosis. NSCLC large cell carcinoma lacks the cytologic, architectural, and histochemical features of small cell carcinoma, adenocarcinoma, or squamous cell carcinoma and is undifferentiated.
When the NSCLC histologic subtype is determined, molecular testing should be performed as part of broad molecular profiling with the goal of identifying rare driver mutations for which effective drugs may already be available or to appropriately counsel patients regarding the availability of clinical trials. NSCLC diagnostic standards include the detection of EGFR, BRAF, and MET mutations, ERBB2 (HER2) expression, and the analysis of ALK, ROS1, RET, and NTRK translocations. In addition, analysis of programmed death-ligand 1 expression is necessary to identify patients who may benefit from the use of immune checkpoint inhibitors.
Surgery combined with chemotherapy has been shown to improve the prognosis of patients with NSCLC large cell carcinoma. Preferred regimens in various lines of treatment and according to molecular characteristics can be found in the NCCN guidelines.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of non–small cell lung cancer (NSCLC) large cell carcinoma.
Lung cancer is the most common cancer worldwide and has the highest mortality rate of all cancers. It comprises two major subtypes: NSCLC and small cell lung cancer (SCLC). Histologically, NSCLC is further classified as adenocarcinoma, squamous cell carcinoma, and large cell carcinoma with or without neuroendocrine features. Large cell carcinoma accounts for 9% of all cases and is frequently associated with poor prognosis. Most patients with NSCLC large cell carcinoma are older than 60 years and are diagnosed with stage III or IV disease. NSCLC large cell carcinoma appears to occur more commonly in men than in women and in patients with a history of smoking. It often presents as a large mass with central necrosis.
NSCLC is often asymptomatic in its early stages. The most frequently reported signs and symptoms of lung cancer include:
• Cough
• Chest pain
• Shortness of breath
• Coughing up blood
• Wheezing
• Hoarseness
• Recurring infections, such as bronchitis and pneumonia
• Weight loss and loss of appetite
• Fatigue
Signs and symptoms of metastatic disease may include bone pain, spinal cord impingement, or neurologic problems, such as headache, weakness or numbness of limbs, dizziness, and seizures.
All patients with NSCLC require a complete staging workup to evaluate the extent of disease because stage plays a central role in treatment selection. After physical examination and a complete blood count, a chest radiograph is often the first test performed. Chest radiographs may show a pulmonary nodule, mass, or infiltrate; mediastinal widening; atelectasis; hilar enlargement; and/or pleural effusion.
Various methods are available to confirm the diagnosis, and the method chosen may be determined at least in part by lesion location. These include:
• Bronchoscopy
• Sputum cytology
• Mediastinoscopy
• Thoracentesis
• Thoracoscopy
• Transthoracic needle biopsy (CT- or fluoroscopy-guided)
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the diagnosis of NSCLC large cell carcinoma requires a thoroughly sampled resected tumor with immunohistochemical stains that exclude adenocarcinoma (TTF-1, napsin A) and squamous cell (p40, p63) carcinoma. Nonresected specimens or cytology specimens are insufficient for its diagnosis. NSCLC large cell carcinoma lacks the cytologic, architectural, and histochemical features of small cell carcinoma, adenocarcinoma, or squamous cell carcinoma and is undifferentiated.
When the NSCLC histologic subtype is determined, molecular testing should be performed as part of broad molecular profiling with the goal of identifying rare driver mutations for which effective drugs may already be available or to appropriately counsel patients regarding the availability of clinical trials. NSCLC diagnostic standards include the detection of EGFR, BRAF, and MET mutations, ERBB2 (HER2) expression, and the analysis of ALK, ROS1, RET, and NTRK translocations. In addition, analysis of programmed death-ligand 1 expression is necessary to identify patients who may benefit from the use of immune checkpoint inhibitors.
Surgery combined with chemotherapy has been shown to improve the prognosis of patients with NSCLC large cell carcinoma. Preferred regimens in various lines of treatment and according to molecular characteristics can be found in the NCCN guidelines.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of non–small cell lung cancer (NSCLC) large cell carcinoma.
Lung cancer is the most common cancer worldwide and has the highest mortality rate of all cancers. It comprises two major subtypes: NSCLC and small cell lung cancer (SCLC). Histologically, NSCLC is further classified as adenocarcinoma, squamous cell carcinoma, and large cell carcinoma with or without neuroendocrine features. Large cell carcinoma accounts for 9% of all cases and is frequently associated with poor prognosis. Most patients with NSCLC large cell carcinoma are older than 60 years and are diagnosed with stage III or IV disease. NSCLC large cell carcinoma appears to occur more commonly in men than in women and in patients with a history of smoking. It often presents as a large mass with central necrosis.
NSCLC is often asymptomatic in its early stages. The most frequently reported signs and symptoms of lung cancer include:
• Cough
• Chest pain
• Shortness of breath
• Coughing up blood
• Wheezing
• Hoarseness
• Recurring infections, such as bronchitis and pneumonia
• Weight loss and loss of appetite
• Fatigue
Signs and symptoms of metastatic disease may include bone pain, spinal cord impingement, or neurologic problems, such as headache, weakness or numbness of limbs, dizziness, and seizures.
All patients with NSCLC require a complete staging workup to evaluate the extent of disease because stage plays a central role in treatment selection. After physical examination and a complete blood count, a chest radiograph is often the first test performed. Chest radiographs may show a pulmonary nodule, mass, or infiltrate; mediastinal widening; atelectasis; hilar enlargement; and/or pleural effusion.
Various methods are available to confirm the diagnosis, and the method chosen may be determined at least in part by lesion location. These include:
• Bronchoscopy
• Sputum cytology
• Mediastinoscopy
• Thoracentesis
• Thoracoscopy
• Transthoracic needle biopsy (CT- or fluoroscopy-guided)
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), the diagnosis of NSCLC large cell carcinoma requires a thoroughly sampled resected tumor with immunohistochemical stains that exclude adenocarcinoma (TTF-1, napsin A) and squamous cell (p40, p63) carcinoma. Nonresected specimens or cytology specimens are insufficient for its diagnosis. NSCLC large cell carcinoma lacks the cytologic, architectural, and histochemical features of small cell carcinoma, adenocarcinoma, or squamous cell carcinoma and is undifferentiated.
When the NSCLC histologic subtype is determined, molecular testing should be performed as part of broad molecular profiling with the goal of identifying rare driver mutations for which effective drugs may already be available or to appropriately counsel patients regarding the availability of clinical trials. NSCLC diagnostic standards include the detection of EGFR, BRAF, and MET mutations, ERBB2 (HER2) expression, and the analysis of ALK, ROS1, RET, and NTRK translocations. In addition, analysis of programmed death-ligand 1 expression is necessary to identify patients who may benefit from the use of immune checkpoint inhibitors.
Surgery combined with chemotherapy has been shown to improve the prognosis of patients with NSCLC large cell carcinoma. Preferred regimens in various lines of treatment and according to molecular characteristics can be found in the NCCN guidelines.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
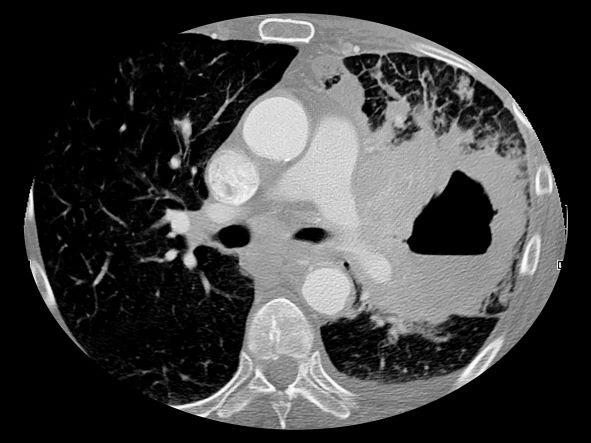
A 67-year-old White man presents to the emergency department with reports of cough, dyspnea, fatigue, hoarseness, and unintentional weight loss. The patient states that his symptoms began approximately 3 weeks earlier and have progressively worsened. In the past year, he has been treated twice for respiratory infections (bronchitis and pneumonia approximately 6 and 9 months before the current presentation, respectively). He has a 45-year history of smoking (45 pack-years). The patient's vital signs include temperature of 100.4 °F, blood pressure of 142/80 mm Hg, and pulse ox of 95%. Physical examination reveals rales in the left side of the chest and decreased breath sounds in bilateral bases of the lungs. The patient appears cachexic. He is 6 ft 2 in and weighs 163 lb.
A chest radiograph reveals a large mass in the left lung field. A subsequent CT of the chest reveals encasement of the left upper and lower lobe bronchus with extensive mediastinal lymphadenopathy and areas of necrosis. Immunohistochemical analysis of the resected tumor reveals a malignant, poorly differentiated epithelial neoplasm composed of large, atypical cells. There is no morphologic or immunohistochemical evidence of glandular, squamous, or neuroendocrine differentiation.
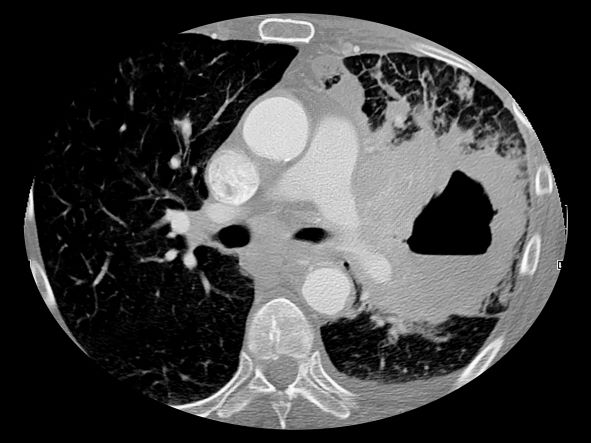
Older woman presents with shortness of breath
Small cell lung cancer (SCLC) is the most aggressive lung cancer subtype, has a poor prognosis, and is highly associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. SCLCs usually grow rapidly and metastasize to mediastinal lymph nodes relatively early in the course of the disease. At presentation, patients may have very large intrathoracic tumors, and distinguishing the primary tumor from lymph node metastases may be difficult.
CT of all common sites of metastasis should be performed to stage the disease. CT scanning of the thorax (lungs and mediastinum) and commonly involved abdominal viscera is the minimum requirement in standard staging workup of SCLC. Intravenous contrast agents should be used whenever possible. In the United States, CT scans of the chest and upper abdomen to include the liver and adrenal glands are standard. The liver is a common site of metastasis.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that these patients be managed with aggressive chemoradiation therapy; in some, lobectomy associated with mediastinal lymph node dissection may be performed. The NCCN notes that advanced disease is managed primarily with chemotherapy, mainly for palliation and symptom control. Older patients, such as the current patient, who have a good performance status (ECOG 0 or 1) and intact organ function should receive standard carboplatin-based chemotherapy. However, even those who have poor prognostic factors (eg, poor performance status, medically significant concomitant conditions) may still be considered for chemotherapy if appropriate precautions are taken to avoid excessive toxicity and further decline in performance status.
Unlike non-SCLC, which has seen waves of new drug approvals from the US Food and Drug Administration (FDA), the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, the FDA approved atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Further, the approval of durvalumab in combination with chemotherapy; and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts
Karl J. D'Silva, MD, has disclosed no relevant financial relationships
Small cell lung cancer (SCLC) is the most aggressive lung cancer subtype, has a poor prognosis, and is highly associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. SCLCs usually grow rapidly and metastasize to mediastinal lymph nodes relatively early in the course of the disease. At presentation, patients may have very large intrathoracic tumors, and distinguishing the primary tumor from lymph node metastases may be difficult.
CT of all common sites of metastasis should be performed to stage the disease. CT scanning of the thorax (lungs and mediastinum) and commonly involved abdominal viscera is the minimum requirement in standard staging workup of SCLC. Intravenous contrast agents should be used whenever possible. In the United States, CT scans of the chest and upper abdomen to include the liver and adrenal glands are standard. The liver is a common site of metastasis.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that these patients be managed with aggressive chemoradiation therapy; in some, lobectomy associated with mediastinal lymph node dissection may be performed. The NCCN notes that advanced disease is managed primarily with chemotherapy, mainly for palliation and symptom control. Older patients, such as the current patient, who have a good performance status (ECOG 0 or 1) and intact organ function should receive standard carboplatin-based chemotherapy. However, even those who have poor prognostic factors (eg, poor performance status, medically significant concomitant conditions) may still be considered for chemotherapy if appropriate precautions are taken to avoid excessive toxicity and further decline in performance status.
Unlike non-SCLC, which has seen waves of new drug approvals from the US Food and Drug Administration (FDA), the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, the FDA approved atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Further, the approval of durvalumab in combination with chemotherapy; and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts
Karl J. D'Silva, MD, has disclosed no relevant financial relationships
Small cell lung cancer (SCLC) is the most aggressive lung cancer subtype, has a poor prognosis, and is highly associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. SCLCs usually grow rapidly and metastasize to mediastinal lymph nodes relatively early in the course of the disease. At presentation, patients may have very large intrathoracic tumors, and distinguishing the primary tumor from lymph node metastases may be difficult.
CT of all common sites of metastasis should be performed to stage the disease. CT scanning of the thorax (lungs and mediastinum) and commonly involved abdominal viscera is the minimum requirement in standard staging workup of SCLC. Intravenous contrast agents should be used whenever possible. In the United States, CT scans of the chest and upper abdomen to include the liver and adrenal glands are standard. The liver is a common site of metastasis.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that these patients be managed with aggressive chemoradiation therapy; in some, lobectomy associated with mediastinal lymph node dissection may be performed. The NCCN notes that advanced disease is managed primarily with chemotherapy, mainly for palliation and symptom control. Older patients, such as the current patient, who have a good performance status (ECOG 0 or 1) and intact organ function should receive standard carboplatin-based chemotherapy. However, even those who have poor prognostic factors (eg, poor performance status, medically significant concomitant conditions) may still be considered for chemotherapy if appropriate precautions are taken to avoid excessive toxicity and further decline in performance status.
Unlike non-SCLC, which has seen waves of new drug approvals from the US Food and Drug Administration (FDA), the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, the FDA approved atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Further, the approval of durvalumab in combination with chemotherapy; and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts
Karl J. D'Silva, MD, has disclosed no relevant financial relationships
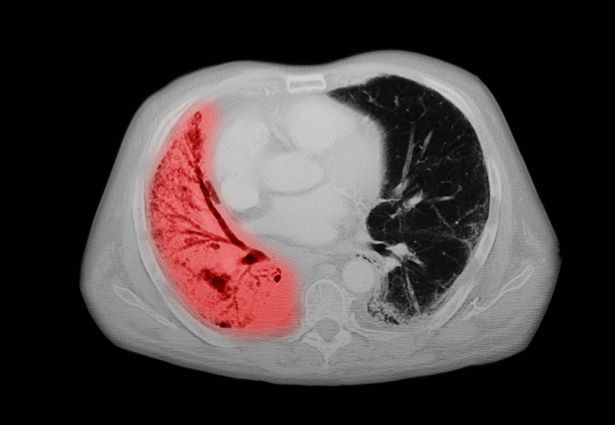
A 72-year-old woman presents with shortness of breath, a productive cough, chest pain, some fatigue, anorexia, a recent 18-pound weight loss, and a history of hypertension. She is currently a smoker and has a 45–pack-year smoking history. On physical examination she has some dullness to percussion with some decreased breath sounds. She has a distended abdomen and complains of itchy skin. The chest x-ray shows a left hilar mass and a 5.4-cm left upper-lobe mass. A CT scan reveals a hilar mass with a mediastinal extension.
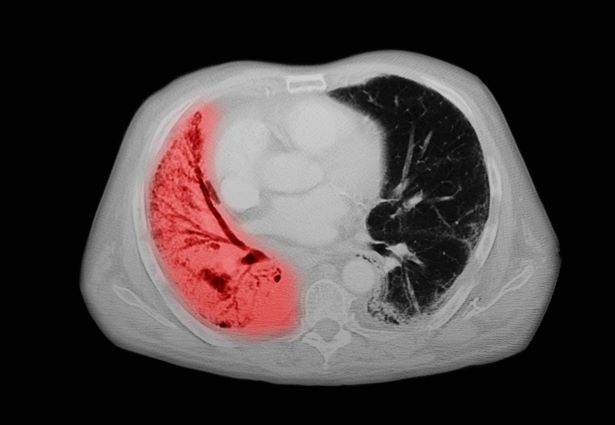
Shortness of breath and chest pain
Liver biopsy will reveal the cause of the liver dysfunction and confirm whether this patient has small cell lung cancer (SCLC), as suspected, and whether it has spread to the liver. This will affect chemotherapy options.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that patients with advanced disease be managed with aggressive chemoradiation therapy and immunotherapy.
SCLC is the most aggressive lung cancer subtype. It is almost always associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. About 70% of patients show metastatic spread at presentation, and the liver is one of the most common sites of metastasis.
Unlike non-SCLC, for which the prognosis has improved in part because of several new drug approvals from the US Food and Drug Administration, the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, with immunotherapy, the outlook may be improving. The NCCN recommends atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Furthermore, the approval of durvalumab in combination with chemotherapy and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with advanced SCLC.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grants from: AstraZeneca; Novis; GSK; Merck
Liver biopsy will reveal the cause of the liver dysfunction and confirm whether this patient has small cell lung cancer (SCLC), as suspected, and whether it has spread to the liver. This will affect chemotherapy options.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that patients with advanced disease be managed with aggressive chemoradiation therapy and immunotherapy.
SCLC is the most aggressive lung cancer subtype. It is almost always associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. About 70% of patients show metastatic spread at presentation, and the liver is one of the most common sites of metastasis.
Unlike non-SCLC, for which the prognosis has improved in part because of several new drug approvals from the US Food and Drug Administration, the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, with immunotherapy, the outlook may be improving. The NCCN recommends atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Furthermore, the approval of durvalumab in combination with chemotherapy and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with advanced SCLC.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grants from: AstraZeneca; Novis; GSK; Merck
Liver biopsy will reveal the cause of the liver dysfunction and confirm whether this patient has small cell lung cancer (SCLC), as suspected, and whether it has spread to the liver. This will affect chemotherapy options.
Most cases of SCLC will present in advanced stages, be inoperable, and have a dismal prognosis. Only about 5% of patients present at an early stage (Ia, Ib, or IIa). The National Comprehensive Cancer Network (NCCN) guidelines recommend that patients with advanced disease be managed with aggressive chemoradiation therapy and immunotherapy.
SCLC is the most aggressive lung cancer subtype. It is almost always associated with smoking. It accounts for 10%-15% of all lung cancers. Rapid tumor growth may lead to obstruction of major airways, with distal collapse leading to postobstructive pneumonitis, infection, and fever. About 70% of patients show metastatic spread at presentation, and the liver is one of the most common sites of metastasis.
Unlike non-SCLC, for which the prognosis has improved in part because of several new drug approvals from the US Food and Drug Administration, the prognosis for SCLC has not changed substantially in the past two decades and remains poor. However, with immunotherapy, the outlook may be improving. The NCCN recommends atezolizumab in combination with carboplatin and etoposide for first-line treatment of adult patients with extensive-stage SCLC. Furthermore, the approval of durvalumab in combination with chemotherapy and the approval of lurbinectedin, a novel chemotherapy agent approved for second-line treatment of SCLC, have added to the therapeutic options for patients with advanced SCLC.
Maurie Markman, MD, President, Department of Medical Oncology, Cancer Treatment Centers of America.
Maurie Markman, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Merck
Serve(d) as a speaker or a member of a speakers bureau for: AstraZeneca; Novis; Glaxo Smith Kline
Received research grants from: AstraZeneca; Novis; GSK; Merck
A 64-year-old man presents with shortness of breath, a productive cough, chest pain, some fatigue, anorexia, a recent 15-lb weight loss, jaundice, and a history of type 2 diabetes. He is a college professor and has a 35–pack-year smoking history; he quit 3 years ago.
On physical examination, he has dullness to percussion and decreased breath sounds. His laboratory data reveal elevated serum liver enzyme levels. Chest radiography shows a left hilar mass and a 5.6-cm left upper lobe mass. CT reveals a hilar mass with a bilateral mediastinal extension and pneumonia. PET shows activity in the left upper lobe mass, with supraclavicular nodal areas and liver lesions. MRI shows that there are no metastases to the brain.