User login
Tremors and memory loss precede Parkinson’s in diverse population
Tremors and memory symptoms were identified among individuals in a primary care setting as early as 10 years before a Parkinson’s disease diagnosis in a new study.
Most research on the causes and early signs of Parkinson’s disease (PD) have involved patients of Northern European ancestry, Cristina Simonet, MD, of Queen Mary University of London, and colleagues wrote in their paper, published in JAMA Neurology.
Additionally, data on how PD might manifest in different ethnic groups are limited, they said.
In their nested case-control, the researchers examined data from electronic health records of an ethnically diverse population of 1,016,277 adults seen in primary care practices between 1990 and Feb. 6, 2018. They compared individuals with PD with those without PD or other neurologic conditions.
The researchers identified 10 age and sex-matched controls for each PD case, and also conducted an unmatched analysis after adjusting for age and sex. The final study population included 1,055 patients with PD and 1,009,523 controls. The population of PD cases was 15.7% Black, 19.7% South Asian, 50.9% White, and 8.3% other; the population of controls was 13.3% Black, 21.5% South Asian, 43.7% White, and 11.3% other.
“We observed a constellation of symptoms noted by general practitioners up to a decade before diagnosis of PD,” the researchers said. Symptoms were identified across three time intervals (less than 2 years, 2-5 years, and 5-10 years before diagnosis) to better evaluate exposure outcome associations.
In the matched analysis of midlife risk factors, epilepsy showed the strongest association with PD diagnosis across all time periods, and type 2 diabetes or hypertension 5-10 years before diagnosis was associated with later PD.
Prediagnostic signs of PD included both motor and nonmotor manifestations.
The matched analysis revealed a significant increased association between tremor and memory symptoms less than 2 years before diagnosis (adjusted odds ratios of 151.24 and 8.73, respectively) as well as up to 10 years before diagnosis for tremors and up to 5 years for memory symptoms (aOR, 11.4 and 3.09, respectively) in PD patients, compared with controls.
Other strong associations between PD and early nonmotor features in cases, compared with controls, included hypotension (aOR, 6.81), constipation (aOR, 3.29), and depression (aOR, 4.61).
In addition, the researchers found associations for epilepsy that had not been identified in previous studies, and these associations persisted in a replication analysis.
The study findings were limited by several factors, mainly the use of routine primary care data with underascertained factors of interest, and potential mislabeling of PD, the researchers noted. Other limitations included the lack of data on prescription medication for PD, and the recording of memory problems in primary care without supportive testing to confirm cognitive impairment.
The results support a range of comorbidities and symptoms that may present in primary care, and clinicians should consider PD as a possible cause, the researchers wrote.
Make early referral a priority
The study is important because of the lack of diversity in Parkinson’s disease research, lead author Dr. Simonet said in an interview.
“Over the last decade, the global population suffering from Parkinson’s disease has more than doubled,” she said. Causes may include the increasing numbers of older people with longer life expectancy. “However, it seems there are other factors, including environmental, genetic, and lifestyle, that might play a role in increasing the prevalence of Parkinson’s disease.”
“More representative studies, including minority ethnic groups and those living in areas of high social and economic deprivation, are needed,” Dr. Simonet emphasized.
She said that there is little research on the association with epilepsy and hearing loss in early PD, and “for that reason, our results should encourage further studies to confirm a possible link between these manifestations and Parkinson’s disease.”
Early detection may drive better diagnoses
The current study is important for understanding the prediagnostic features and risk factors that may allow for earlier detection of Parkinson’s disease, William Hung, MD, a geriatrics and palliative care specialist of the Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Prior to this study, there was limited understanding of these features.
“One surprise [in the findings] was that ethnicity and socioeconomic deprivation do not appear to be associated with the risk of PD, in contrast to other illnesses such as dementia,” said Dr. Hung. “The array of prediagnostic features associated with PD is not surprising, but nonetheless important for clinicians to know to consider whether PD could be the underlying cause.”
The take-home message for primary care is that “there are features, such as hearing loss, history of epilepsy, autonomic symptoms, motor symptoms, among others, for which clinicians should consider PD as part of the differential diagnosis as underlying cause and consider referral to specialists for diagnostic clarification,” said Dr. Hung.
“Additional research is needed to translate these findings to care, perhaps developing decision aids, interventions that may help with diagnosis and evaluation,” as is work on understanding the link between PD and symptoms such as hearing loss and epilepsy, he said.
Primary care offers opportunity to identify risk factors
The current study represents an important step in early recognition of PD, with implications for helping patients access treatments promptly and improve their quality of life, Bhavana Patel, DO, Shannon Chiu, MD, and Melissa J. Armstrong, MD, of the University of Florida, Gainesville, wrote in an accompanying editorial.
“The primary care setting is commonly where symptoms heralding the onset of PD are first discussed. However, little is known regarding the prediagnostic manifestations of PD that are seen in primary care clinics, particularly in underserved populations,” they wrote.
The study included many risk factors and prodromal markers associated with research criteria for prodromal PD, but did not include several risk and prodromal markers in the Movement Disorders Society research criteria, “such as symptoms suggestive of REM sleep behavior disorder, excessive daytime sleepiness (which overlaps with, but is distinct from, fatigue), urinary dysfunction, pesticide and solvent exposure, caffeine use, level of physical activity, and family history,” they said.
Even in individuals with diagnosed PD, certain symptoms, particularly nonmotor symptoms, are commonly underreported,” and primary care clinicians may not recognize these symptoms as PD risk factors, the authors noted.
However, “in addition to contributing to possible models of modifiable risk factors for PD, study results may also further inform algorithms designed to predict PD diagnoses in primary care,” they said. The study also highlights the need for more multivariable models to better identify PD risk factors and strategies for early identification of PD in primary care.
Several study coauthors received funding related to the study from Barts Charity, Health Data Research UK, the Department of Health and Social Care (England) and the devolved administrations, and leading medical research charities, as well as the National Institute for Health Research UCLH Biomedical Research Centre. Lead author Dr. Simonet and Dr. Hung had no financial conflicts to disclose. Dr. Patel disclosed support from the National Institute on Aging, the Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia, and the American Brain Foundation and the Mary E. Groff Charitable Trust. Dr. Chiu reported receiving grants from Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia and the Smallwood Foundation. Dr. Armstrong disclosed funding from the National Institute on Aging, the Florida Department of Health, the Lewy Body Dementia Association, the Alzheimer’s Therapeutic Research Institute/Alzheimer’s Clinical Trial Consortium, the Alzheimer’s Disease Cooperative Study as Data Safety Monitoring Board the Parkinson’s Foundation, and the American Academy of Neurology.
Tremors and memory symptoms were identified among individuals in a primary care setting as early as 10 years before a Parkinson’s disease diagnosis in a new study.
Most research on the causes and early signs of Parkinson’s disease (PD) have involved patients of Northern European ancestry, Cristina Simonet, MD, of Queen Mary University of London, and colleagues wrote in their paper, published in JAMA Neurology.
Additionally, data on how PD might manifest in different ethnic groups are limited, they said.
In their nested case-control, the researchers examined data from electronic health records of an ethnically diverse population of 1,016,277 adults seen in primary care practices between 1990 and Feb. 6, 2018. They compared individuals with PD with those without PD or other neurologic conditions.
The researchers identified 10 age and sex-matched controls for each PD case, and also conducted an unmatched analysis after adjusting for age and sex. The final study population included 1,055 patients with PD and 1,009,523 controls. The population of PD cases was 15.7% Black, 19.7% South Asian, 50.9% White, and 8.3% other; the population of controls was 13.3% Black, 21.5% South Asian, 43.7% White, and 11.3% other.
“We observed a constellation of symptoms noted by general practitioners up to a decade before diagnosis of PD,” the researchers said. Symptoms were identified across three time intervals (less than 2 years, 2-5 years, and 5-10 years before diagnosis) to better evaluate exposure outcome associations.
In the matched analysis of midlife risk factors, epilepsy showed the strongest association with PD diagnosis across all time periods, and type 2 diabetes or hypertension 5-10 years before diagnosis was associated with later PD.
Prediagnostic signs of PD included both motor and nonmotor manifestations.
The matched analysis revealed a significant increased association between tremor and memory symptoms less than 2 years before diagnosis (adjusted odds ratios of 151.24 and 8.73, respectively) as well as up to 10 years before diagnosis for tremors and up to 5 years for memory symptoms (aOR, 11.4 and 3.09, respectively) in PD patients, compared with controls.
Other strong associations between PD and early nonmotor features in cases, compared with controls, included hypotension (aOR, 6.81), constipation (aOR, 3.29), and depression (aOR, 4.61).
In addition, the researchers found associations for epilepsy that had not been identified in previous studies, and these associations persisted in a replication analysis.
The study findings were limited by several factors, mainly the use of routine primary care data with underascertained factors of interest, and potential mislabeling of PD, the researchers noted. Other limitations included the lack of data on prescription medication for PD, and the recording of memory problems in primary care without supportive testing to confirm cognitive impairment.
The results support a range of comorbidities and symptoms that may present in primary care, and clinicians should consider PD as a possible cause, the researchers wrote.
Make early referral a priority
The study is important because of the lack of diversity in Parkinson’s disease research, lead author Dr. Simonet said in an interview.
“Over the last decade, the global population suffering from Parkinson’s disease has more than doubled,” she said. Causes may include the increasing numbers of older people with longer life expectancy. “However, it seems there are other factors, including environmental, genetic, and lifestyle, that might play a role in increasing the prevalence of Parkinson’s disease.”
“More representative studies, including minority ethnic groups and those living in areas of high social and economic deprivation, are needed,” Dr. Simonet emphasized.
She said that there is little research on the association with epilepsy and hearing loss in early PD, and “for that reason, our results should encourage further studies to confirm a possible link between these manifestations and Parkinson’s disease.”
Early detection may drive better diagnoses
The current study is important for understanding the prediagnostic features and risk factors that may allow for earlier detection of Parkinson’s disease, William Hung, MD, a geriatrics and palliative care specialist of the Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Prior to this study, there was limited understanding of these features.
“One surprise [in the findings] was that ethnicity and socioeconomic deprivation do not appear to be associated with the risk of PD, in contrast to other illnesses such as dementia,” said Dr. Hung. “The array of prediagnostic features associated with PD is not surprising, but nonetheless important for clinicians to know to consider whether PD could be the underlying cause.”
The take-home message for primary care is that “there are features, such as hearing loss, history of epilepsy, autonomic symptoms, motor symptoms, among others, for which clinicians should consider PD as part of the differential diagnosis as underlying cause and consider referral to specialists for diagnostic clarification,” said Dr. Hung.
“Additional research is needed to translate these findings to care, perhaps developing decision aids, interventions that may help with diagnosis and evaluation,” as is work on understanding the link between PD and symptoms such as hearing loss and epilepsy, he said.
Primary care offers opportunity to identify risk factors
The current study represents an important step in early recognition of PD, with implications for helping patients access treatments promptly and improve their quality of life, Bhavana Patel, DO, Shannon Chiu, MD, and Melissa J. Armstrong, MD, of the University of Florida, Gainesville, wrote in an accompanying editorial.
“The primary care setting is commonly where symptoms heralding the onset of PD are first discussed. However, little is known regarding the prediagnostic manifestations of PD that are seen in primary care clinics, particularly in underserved populations,” they wrote.
The study included many risk factors and prodromal markers associated with research criteria for prodromal PD, but did not include several risk and prodromal markers in the Movement Disorders Society research criteria, “such as symptoms suggestive of REM sleep behavior disorder, excessive daytime sleepiness (which overlaps with, but is distinct from, fatigue), urinary dysfunction, pesticide and solvent exposure, caffeine use, level of physical activity, and family history,” they said.
Even in individuals with diagnosed PD, certain symptoms, particularly nonmotor symptoms, are commonly underreported,” and primary care clinicians may not recognize these symptoms as PD risk factors, the authors noted.
However, “in addition to contributing to possible models of modifiable risk factors for PD, study results may also further inform algorithms designed to predict PD diagnoses in primary care,” they said. The study also highlights the need for more multivariable models to better identify PD risk factors and strategies for early identification of PD in primary care.
Several study coauthors received funding related to the study from Barts Charity, Health Data Research UK, the Department of Health and Social Care (England) and the devolved administrations, and leading medical research charities, as well as the National Institute for Health Research UCLH Biomedical Research Centre. Lead author Dr. Simonet and Dr. Hung had no financial conflicts to disclose. Dr. Patel disclosed support from the National Institute on Aging, the Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia, and the American Brain Foundation and the Mary E. Groff Charitable Trust. Dr. Chiu reported receiving grants from Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia and the Smallwood Foundation. Dr. Armstrong disclosed funding from the National Institute on Aging, the Florida Department of Health, the Lewy Body Dementia Association, the Alzheimer’s Therapeutic Research Institute/Alzheimer’s Clinical Trial Consortium, the Alzheimer’s Disease Cooperative Study as Data Safety Monitoring Board the Parkinson’s Foundation, and the American Academy of Neurology.
Tremors and memory symptoms were identified among individuals in a primary care setting as early as 10 years before a Parkinson’s disease diagnosis in a new study.
Most research on the causes and early signs of Parkinson’s disease (PD) have involved patients of Northern European ancestry, Cristina Simonet, MD, of Queen Mary University of London, and colleagues wrote in their paper, published in JAMA Neurology.
Additionally, data on how PD might manifest in different ethnic groups are limited, they said.
In their nested case-control, the researchers examined data from electronic health records of an ethnically diverse population of 1,016,277 adults seen in primary care practices between 1990 and Feb. 6, 2018. They compared individuals with PD with those without PD or other neurologic conditions.
The researchers identified 10 age and sex-matched controls for each PD case, and also conducted an unmatched analysis after adjusting for age and sex. The final study population included 1,055 patients with PD and 1,009,523 controls. The population of PD cases was 15.7% Black, 19.7% South Asian, 50.9% White, and 8.3% other; the population of controls was 13.3% Black, 21.5% South Asian, 43.7% White, and 11.3% other.
“We observed a constellation of symptoms noted by general practitioners up to a decade before diagnosis of PD,” the researchers said. Symptoms were identified across three time intervals (less than 2 years, 2-5 years, and 5-10 years before diagnosis) to better evaluate exposure outcome associations.
In the matched analysis of midlife risk factors, epilepsy showed the strongest association with PD diagnosis across all time periods, and type 2 diabetes or hypertension 5-10 years before diagnosis was associated with later PD.
Prediagnostic signs of PD included both motor and nonmotor manifestations.
The matched analysis revealed a significant increased association between tremor and memory symptoms less than 2 years before diagnosis (adjusted odds ratios of 151.24 and 8.73, respectively) as well as up to 10 years before diagnosis for tremors and up to 5 years for memory symptoms (aOR, 11.4 and 3.09, respectively) in PD patients, compared with controls.
Other strong associations between PD and early nonmotor features in cases, compared with controls, included hypotension (aOR, 6.81), constipation (aOR, 3.29), and depression (aOR, 4.61).
In addition, the researchers found associations for epilepsy that had not been identified in previous studies, and these associations persisted in a replication analysis.
The study findings were limited by several factors, mainly the use of routine primary care data with underascertained factors of interest, and potential mislabeling of PD, the researchers noted. Other limitations included the lack of data on prescription medication for PD, and the recording of memory problems in primary care without supportive testing to confirm cognitive impairment.
The results support a range of comorbidities and symptoms that may present in primary care, and clinicians should consider PD as a possible cause, the researchers wrote.
Make early referral a priority
The study is important because of the lack of diversity in Parkinson’s disease research, lead author Dr. Simonet said in an interview.
“Over the last decade, the global population suffering from Parkinson’s disease has more than doubled,” she said. Causes may include the increasing numbers of older people with longer life expectancy. “However, it seems there are other factors, including environmental, genetic, and lifestyle, that might play a role in increasing the prevalence of Parkinson’s disease.”
“More representative studies, including minority ethnic groups and those living in areas of high social and economic deprivation, are needed,” Dr. Simonet emphasized.
She said that there is little research on the association with epilepsy and hearing loss in early PD, and “for that reason, our results should encourage further studies to confirm a possible link between these manifestations and Parkinson’s disease.”
Early detection may drive better diagnoses
The current study is important for understanding the prediagnostic features and risk factors that may allow for earlier detection of Parkinson’s disease, William Hung, MD, a geriatrics and palliative care specialist of the Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Prior to this study, there was limited understanding of these features.
“One surprise [in the findings] was that ethnicity and socioeconomic deprivation do not appear to be associated with the risk of PD, in contrast to other illnesses such as dementia,” said Dr. Hung. “The array of prediagnostic features associated with PD is not surprising, but nonetheless important for clinicians to know to consider whether PD could be the underlying cause.”
The take-home message for primary care is that “there are features, such as hearing loss, history of epilepsy, autonomic symptoms, motor symptoms, among others, for which clinicians should consider PD as part of the differential diagnosis as underlying cause and consider referral to specialists for diagnostic clarification,” said Dr. Hung.
“Additional research is needed to translate these findings to care, perhaps developing decision aids, interventions that may help with diagnosis and evaluation,” as is work on understanding the link between PD and symptoms such as hearing loss and epilepsy, he said.
Primary care offers opportunity to identify risk factors
The current study represents an important step in early recognition of PD, with implications for helping patients access treatments promptly and improve their quality of life, Bhavana Patel, DO, Shannon Chiu, MD, and Melissa J. Armstrong, MD, of the University of Florida, Gainesville, wrote in an accompanying editorial.
“The primary care setting is commonly where symptoms heralding the onset of PD are first discussed. However, little is known regarding the prediagnostic manifestations of PD that are seen in primary care clinics, particularly in underserved populations,” they wrote.
The study included many risk factors and prodromal markers associated with research criteria for prodromal PD, but did not include several risk and prodromal markers in the Movement Disorders Society research criteria, “such as symptoms suggestive of REM sleep behavior disorder, excessive daytime sleepiness (which overlaps with, but is distinct from, fatigue), urinary dysfunction, pesticide and solvent exposure, caffeine use, level of physical activity, and family history,” they said.
Even in individuals with diagnosed PD, certain symptoms, particularly nonmotor symptoms, are commonly underreported,” and primary care clinicians may not recognize these symptoms as PD risk factors, the authors noted.
However, “in addition to contributing to possible models of modifiable risk factors for PD, study results may also further inform algorithms designed to predict PD diagnoses in primary care,” they said. The study also highlights the need for more multivariable models to better identify PD risk factors and strategies for early identification of PD in primary care.
Several study coauthors received funding related to the study from Barts Charity, Health Data Research UK, the Department of Health and Social Care (England) and the devolved administrations, and leading medical research charities, as well as the National Institute for Health Research UCLH Biomedical Research Centre. Lead author Dr. Simonet and Dr. Hung had no financial conflicts to disclose. Dr. Patel disclosed support from the National Institute on Aging, the Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia, and the American Brain Foundation and the Mary E. Groff Charitable Trust. Dr. Chiu reported receiving grants from Mangurian-Fixel-McKnight Collaboration for Pilot Studies in Lewy Body Dementia and the Smallwood Foundation. Dr. Armstrong disclosed funding from the National Institute on Aging, the Florida Department of Health, the Lewy Body Dementia Association, the Alzheimer’s Therapeutic Research Institute/Alzheimer’s Clinical Trial Consortium, the Alzheimer’s Disease Cooperative Study as Data Safety Monitoring Board the Parkinson’s Foundation, and the American Academy of Neurology.
FROM JAMA NEUROLOGY
Long COVID patients may develop nerve damage: Study
according to a new study published in the journal Neurology: Neuroimmunology & Neuroinflammation (doi: 10.1212/NXI.0000000000001146).
The nerve damage, which has been seen even among mild coronavirus cases, appears to be caused by immunity problems triggered by infection.
“This is one of the early papers looking into causes of long COVID, which will steadily increase in importance as acute COVID wanes,” Anne Louise Oaklander, MD, the lead study author and a neurologist at Massachusetts General Hospital, Boston, said in a statement.
“Our findings suggest that some long COVID patients had damage to their peripheral nerve fibers and that damage to the small-fiber type of nerve cell may be prominent,” she said.
The research team analyzed data from 17 COVID-19 survivors with lingering symptoms who had no history or risks of neuropathy, or nerve damage or disease. The patients were from 10 states and territories, and all but one had mild infections.
They found that 10 patients – or 59% – had at least one test that confirmed neuropathy. Two patients had rare neuropathies that affected muscle nerves, and 10 were diagnosed with small-fiber neuropathy, which is a cause of chronic pain. Common symptoms included fatigue, weakness, changes in their senses, and pain in their hands and feet.
For treatment, 11 patients were given immunotherapies such as corticosteroids or intravenous immunoglobulins, and the five patients who received repeated IgG treatments appeared to benefit. Over time, 52% of patients improved, though none had all of their symptoms go away.
“Research from our team and others is clarifying what the different types of post-COVID neuropathy are and how best to diagnose and treat them,” she said. “Most long COVID neuropathies described so far appear to reflect immune responses to the virus that went off course.”
Dr. Oaklander noted that researchers haven’t been able to do clinical trials to evaluate specific post-COVID neuropathy treatments. But some existing treatments may help.
“Some patients seem to improve from standard treatments for other immune-related neuropathies,” she said.
A version of this article first appeared on WebMD.com.
according to a new study published in the journal Neurology: Neuroimmunology & Neuroinflammation (doi: 10.1212/NXI.0000000000001146).
The nerve damage, which has been seen even among mild coronavirus cases, appears to be caused by immunity problems triggered by infection.
“This is one of the early papers looking into causes of long COVID, which will steadily increase in importance as acute COVID wanes,” Anne Louise Oaklander, MD, the lead study author and a neurologist at Massachusetts General Hospital, Boston, said in a statement.
“Our findings suggest that some long COVID patients had damage to their peripheral nerve fibers and that damage to the small-fiber type of nerve cell may be prominent,” she said.
The research team analyzed data from 17 COVID-19 survivors with lingering symptoms who had no history or risks of neuropathy, or nerve damage or disease. The patients were from 10 states and territories, and all but one had mild infections.
They found that 10 patients – or 59% – had at least one test that confirmed neuropathy. Two patients had rare neuropathies that affected muscle nerves, and 10 were diagnosed with small-fiber neuropathy, which is a cause of chronic pain. Common symptoms included fatigue, weakness, changes in their senses, and pain in their hands and feet.
For treatment, 11 patients were given immunotherapies such as corticosteroids or intravenous immunoglobulins, and the five patients who received repeated IgG treatments appeared to benefit. Over time, 52% of patients improved, though none had all of their symptoms go away.
“Research from our team and others is clarifying what the different types of post-COVID neuropathy are and how best to diagnose and treat them,” she said. “Most long COVID neuropathies described so far appear to reflect immune responses to the virus that went off course.”
Dr. Oaklander noted that researchers haven’t been able to do clinical trials to evaluate specific post-COVID neuropathy treatments. But some existing treatments may help.
“Some patients seem to improve from standard treatments for other immune-related neuropathies,” she said.
A version of this article first appeared on WebMD.com.
according to a new study published in the journal Neurology: Neuroimmunology & Neuroinflammation (doi: 10.1212/NXI.0000000000001146).
The nerve damage, which has been seen even among mild coronavirus cases, appears to be caused by immunity problems triggered by infection.
“This is one of the early papers looking into causes of long COVID, which will steadily increase in importance as acute COVID wanes,” Anne Louise Oaklander, MD, the lead study author and a neurologist at Massachusetts General Hospital, Boston, said in a statement.
“Our findings suggest that some long COVID patients had damage to their peripheral nerve fibers and that damage to the small-fiber type of nerve cell may be prominent,” she said.
The research team analyzed data from 17 COVID-19 survivors with lingering symptoms who had no history or risks of neuropathy, or nerve damage or disease. The patients were from 10 states and territories, and all but one had mild infections.
They found that 10 patients – or 59% – had at least one test that confirmed neuropathy. Two patients had rare neuropathies that affected muscle nerves, and 10 were diagnosed with small-fiber neuropathy, which is a cause of chronic pain. Common symptoms included fatigue, weakness, changes in their senses, and pain in their hands and feet.
For treatment, 11 patients were given immunotherapies such as corticosteroids or intravenous immunoglobulins, and the five patients who received repeated IgG treatments appeared to benefit. Over time, 52% of patients improved, though none had all of their symptoms go away.
“Research from our team and others is clarifying what the different types of post-COVID neuropathy are and how best to diagnose and treat them,” she said. “Most long COVID neuropathies described so far appear to reflect immune responses to the virus that went off course.”
Dr. Oaklander noted that researchers haven’t been able to do clinical trials to evaluate specific post-COVID neuropathy treatments. But some existing treatments may help.
“Some patients seem to improve from standard treatments for other immune-related neuropathies,” she said.
A version of this article first appeared on WebMD.com.
FROM NEUROLOGY: NEUROIMMUNOLOGY & NEUROINFLAMMATION
Strep infection and tics in children: new data
Group A streptococcus (GAS) infection is not associated with new-onset tic disorders in at-risk children, findings from a large prospective study show.
The results mean that if preteens present with a new-onset tic condition, “they’re unlikely to have it as a result of a group A streptococcal throat infection,” study author Anette Eleonore Schrag, MD, PhD, professor, department of clinical neuroscience, Institute of Neurology, University College London, told this news organization.
Therefore, clinicians should not automatically prescribe antibiotics for children with tics, which sometimes occurs, said Dr. Schrag.
The study was published online Feb. 2 in Neurology.
Ongoing controversy
Research shows that genetic and environmental factors contribute to chronic tic disorders (CTDs) and Tourette syndrome (TS). Prenatal exposure to maternal smoking and central nervous system (CNS) stimulants, as well as psychosocial stress, may play a role.
There has been an ongoing controversy regarding the possible role of GAS in tics, with some studies showing an association and others not showing a link. However, previous studies have been retrospective, registry based, or had limited sample size.
This new prospective study is the first in children without a tic disorder but who were at relatively high risk of developing one. The children were followed to assess development of streptococcal infections and tics, said Dr. Schrag.
The study included 259 children aged 3-10 years (mean baseline age, 6.8 years; over half female) who had a first-degree relative such as a parent or sibling with TS or CTD.
The average age at TS onset is 7 years, peaking in prevalence and severity at about 9-12 years. GAS throat infections are common in this age group.
Although study participants did not have tics themselves, they represented “an enriched group,” said Dr. Schrag. “Because they had family history, we knew they were at increased risk for developing tics.”
Participants were evaluated every 2 months, alternating between scheduled hospital visits and telephone interviews. Parents kept a weekly diary and were instructed to bring their child in for assessment if they showed any signs of tics.
The average follow-up period was 1.6 years, but some of the children were followed for up to 48 months. During the study, there were a total of 1,944 assessments, including 939 telephone interviews and 1,005 clinical visits.
More common in boys
Investigators defined tic onset as the first occurrence of any sudden, rapid, recurrent, nonrhythmic involuntary movement and/or vocalization on at least three separate days within a period of 3 weeks.
The investigators assessed GAS exposure using parameters from throat swabs, serum anti-streptolysin O titers, and anti-DNAse B titers.
They used multiple definitions and combinations of GAS exposures “to make sure we weren’t missing any association because we didn’t use the right definition,” said Dr. Schrag. She explained a definitive strep infection is not always clear-cut.
At baseline, 17.0% participants tested positive for GAS, and 78.8% tested negative. No throat swab was available from 4.2% of participants.
During follow-up, the number of confirmed positive GAS exposures was 59, 102, 125, and 138, depending on the definition.
Researchers identified 61 tic cases during the study period. There was no evidence of an association of tic onset with GAS exposure after adjusting for age, sex, and parental education level.
However, there was a strong association between tic onset and sex, with girls being 60% less likely to develop tics than boys (hazard ratio, 0.4; 95% CI, 0.2-0.7; P < .01).
This result wasn’t particularly surprising, as it’s known that more boys develop tics than girls. “We just confirmed that in a prospective way,” said Dr. Schrag.
Results from sensitivity analyses confirmed the results. This was also the case with analyses that excluded visits with missing data on GAS exposure and that further adjusted for clinical site and psychotropic medication use.
Other pathogens?
Although the results showed no association between strep and tics in this population, it does not “close the door completely” on a potential relationship, said Dr. Schrag.
“By and large, the development of tics in children is not associated with group A strep, but differences in small subgroups can never be excluded by a study like this.”
Participants in this study were part of the European Multicentre Tics in Children Studies (EMTICS), a prospective cohort study exploring the role of environmental and genetic factors in pediatric CTD. That project is also looking at immune system factors, “which might play a role in the development of chronic tic disorder and associated conditions,” said Dr. Schrag.
It’s still possible, she added, that other pathogens could play a role in tic development. “That’s going to be the subject of further analysis and future studies,” she said.
Tamara Pringsheim, MD, professor of clinical neurosciences, psychiatry, pediatrics, and community health sciences, University of Calgary (Alta.), praised the research.
“This was a well-designed study, with a large sample of 260 children followed for up to 4 years, using a standardized protocol to assess for group A streptococcal infection and new onset of tics.”
The study, which did not uncover an association between GAS exposure and tic onset, “provides high level evidence that group A streptococcal exposure is not an important risk factor for the new onset of tics in children with a family history of tic disorders.”
The study received funding from the European Union Seventh Framework Program for research technological development and demonstration. Dr. Schrag reports receiving consultancy or advisory board honoraria from Biogen, Abbvie, Bial, and Neurotechnology; research support from the National Institute of Health Research, Parkinsons UK, and the Economic and Social Research Council and the European Commission; and Royalties from Oxford University Press. Dr. Pringsheim reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Group A streptococcus (GAS) infection is not associated with new-onset tic disorders in at-risk children, findings from a large prospective study show.
The results mean that if preteens present with a new-onset tic condition, “they’re unlikely to have it as a result of a group A streptococcal throat infection,” study author Anette Eleonore Schrag, MD, PhD, professor, department of clinical neuroscience, Institute of Neurology, University College London, told this news organization.
Therefore, clinicians should not automatically prescribe antibiotics for children with tics, which sometimes occurs, said Dr. Schrag.
The study was published online Feb. 2 in Neurology.
Ongoing controversy
Research shows that genetic and environmental factors contribute to chronic tic disorders (CTDs) and Tourette syndrome (TS). Prenatal exposure to maternal smoking and central nervous system (CNS) stimulants, as well as psychosocial stress, may play a role.
There has been an ongoing controversy regarding the possible role of GAS in tics, with some studies showing an association and others not showing a link. However, previous studies have been retrospective, registry based, or had limited sample size.
This new prospective study is the first in children without a tic disorder but who were at relatively high risk of developing one. The children were followed to assess development of streptococcal infections and tics, said Dr. Schrag.
The study included 259 children aged 3-10 years (mean baseline age, 6.8 years; over half female) who had a first-degree relative such as a parent or sibling with TS or CTD.
The average age at TS onset is 7 years, peaking in prevalence and severity at about 9-12 years. GAS throat infections are common in this age group.
Although study participants did not have tics themselves, they represented “an enriched group,” said Dr. Schrag. “Because they had family history, we knew they were at increased risk for developing tics.”
Participants were evaluated every 2 months, alternating between scheduled hospital visits and telephone interviews. Parents kept a weekly diary and were instructed to bring their child in for assessment if they showed any signs of tics.
The average follow-up period was 1.6 years, but some of the children were followed for up to 48 months. During the study, there were a total of 1,944 assessments, including 939 telephone interviews and 1,005 clinical visits.
More common in boys
Investigators defined tic onset as the first occurrence of any sudden, rapid, recurrent, nonrhythmic involuntary movement and/or vocalization on at least three separate days within a period of 3 weeks.
The investigators assessed GAS exposure using parameters from throat swabs, serum anti-streptolysin O titers, and anti-DNAse B titers.
They used multiple definitions and combinations of GAS exposures “to make sure we weren’t missing any association because we didn’t use the right definition,” said Dr. Schrag. She explained a definitive strep infection is not always clear-cut.
At baseline, 17.0% participants tested positive for GAS, and 78.8% tested negative. No throat swab was available from 4.2% of participants.
During follow-up, the number of confirmed positive GAS exposures was 59, 102, 125, and 138, depending on the definition.
Researchers identified 61 tic cases during the study period. There was no evidence of an association of tic onset with GAS exposure after adjusting for age, sex, and parental education level.
However, there was a strong association between tic onset and sex, with girls being 60% less likely to develop tics than boys (hazard ratio, 0.4; 95% CI, 0.2-0.7; P < .01).
This result wasn’t particularly surprising, as it’s known that more boys develop tics than girls. “We just confirmed that in a prospective way,” said Dr. Schrag.
Results from sensitivity analyses confirmed the results. This was also the case with analyses that excluded visits with missing data on GAS exposure and that further adjusted for clinical site and psychotropic medication use.
Other pathogens?
Although the results showed no association between strep and tics in this population, it does not “close the door completely” on a potential relationship, said Dr. Schrag.
“By and large, the development of tics in children is not associated with group A strep, but differences in small subgroups can never be excluded by a study like this.”
Participants in this study were part of the European Multicentre Tics in Children Studies (EMTICS), a prospective cohort study exploring the role of environmental and genetic factors in pediatric CTD. That project is also looking at immune system factors, “which might play a role in the development of chronic tic disorder and associated conditions,” said Dr. Schrag.
It’s still possible, she added, that other pathogens could play a role in tic development. “That’s going to be the subject of further analysis and future studies,” she said.
Tamara Pringsheim, MD, professor of clinical neurosciences, psychiatry, pediatrics, and community health sciences, University of Calgary (Alta.), praised the research.
“This was a well-designed study, with a large sample of 260 children followed for up to 4 years, using a standardized protocol to assess for group A streptococcal infection and new onset of tics.”
The study, which did not uncover an association between GAS exposure and tic onset, “provides high level evidence that group A streptococcal exposure is not an important risk factor for the new onset of tics in children with a family history of tic disorders.”
The study received funding from the European Union Seventh Framework Program for research technological development and demonstration. Dr. Schrag reports receiving consultancy or advisory board honoraria from Biogen, Abbvie, Bial, and Neurotechnology; research support from the National Institute of Health Research, Parkinsons UK, and the Economic and Social Research Council and the European Commission; and Royalties from Oxford University Press. Dr. Pringsheim reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Group A streptococcus (GAS) infection is not associated with new-onset tic disorders in at-risk children, findings from a large prospective study show.
The results mean that if preteens present with a new-onset tic condition, “they’re unlikely to have it as a result of a group A streptococcal throat infection,” study author Anette Eleonore Schrag, MD, PhD, professor, department of clinical neuroscience, Institute of Neurology, University College London, told this news organization.
Therefore, clinicians should not automatically prescribe antibiotics for children with tics, which sometimes occurs, said Dr. Schrag.
The study was published online Feb. 2 in Neurology.
Ongoing controversy
Research shows that genetic and environmental factors contribute to chronic tic disorders (CTDs) and Tourette syndrome (TS). Prenatal exposure to maternal smoking and central nervous system (CNS) stimulants, as well as psychosocial stress, may play a role.
There has been an ongoing controversy regarding the possible role of GAS in tics, with some studies showing an association and others not showing a link. However, previous studies have been retrospective, registry based, or had limited sample size.
This new prospective study is the first in children without a tic disorder but who were at relatively high risk of developing one. The children were followed to assess development of streptococcal infections and tics, said Dr. Schrag.
The study included 259 children aged 3-10 years (mean baseline age, 6.8 years; over half female) who had a first-degree relative such as a parent or sibling with TS or CTD.
The average age at TS onset is 7 years, peaking in prevalence and severity at about 9-12 years. GAS throat infections are common in this age group.
Although study participants did not have tics themselves, they represented “an enriched group,” said Dr. Schrag. “Because they had family history, we knew they were at increased risk for developing tics.”
Participants were evaluated every 2 months, alternating between scheduled hospital visits and telephone interviews. Parents kept a weekly diary and were instructed to bring their child in for assessment if they showed any signs of tics.
The average follow-up period was 1.6 years, but some of the children were followed for up to 48 months. During the study, there were a total of 1,944 assessments, including 939 telephone interviews and 1,005 clinical visits.
More common in boys
Investigators defined tic onset as the first occurrence of any sudden, rapid, recurrent, nonrhythmic involuntary movement and/or vocalization on at least three separate days within a period of 3 weeks.
The investigators assessed GAS exposure using parameters from throat swabs, serum anti-streptolysin O titers, and anti-DNAse B titers.
They used multiple definitions and combinations of GAS exposures “to make sure we weren’t missing any association because we didn’t use the right definition,” said Dr. Schrag. She explained a definitive strep infection is not always clear-cut.
At baseline, 17.0% participants tested positive for GAS, and 78.8% tested negative. No throat swab was available from 4.2% of participants.
During follow-up, the number of confirmed positive GAS exposures was 59, 102, 125, and 138, depending on the definition.
Researchers identified 61 tic cases during the study period. There was no evidence of an association of tic onset with GAS exposure after adjusting for age, sex, and parental education level.
However, there was a strong association between tic onset and sex, with girls being 60% less likely to develop tics than boys (hazard ratio, 0.4; 95% CI, 0.2-0.7; P < .01).
This result wasn’t particularly surprising, as it’s known that more boys develop tics than girls. “We just confirmed that in a prospective way,” said Dr. Schrag.
Results from sensitivity analyses confirmed the results. This was also the case with analyses that excluded visits with missing data on GAS exposure and that further adjusted for clinical site and psychotropic medication use.
Other pathogens?
Although the results showed no association between strep and tics in this population, it does not “close the door completely” on a potential relationship, said Dr. Schrag.
“By and large, the development of tics in children is not associated with group A strep, but differences in small subgroups can never be excluded by a study like this.”
Participants in this study were part of the European Multicentre Tics in Children Studies (EMTICS), a prospective cohort study exploring the role of environmental and genetic factors in pediatric CTD. That project is also looking at immune system factors, “which might play a role in the development of chronic tic disorder and associated conditions,” said Dr. Schrag.
It’s still possible, she added, that other pathogens could play a role in tic development. “That’s going to be the subject of further analysis and future studies,” she said.
Tamara Pringsheim, MD, professor of clinical neurosciences, psychiatry, pediatrics, and community health sciences, University of Calgary (Alta.), praised the research.
“This was a well-designed study, with a large sample of 260 children followed for up to 4 years, using a standardized protocol to assess for group A streptococcal infection and new onset of tics.”
The study, which did not uncover an association between GAS exposure and tic onset, “provides high level evidence that group A streptococcal exposure is not an important risk factor for the new onset of tics in children with a family history of tic disorders.”
The study received funding from the European Union Seventh Framework Program for research technological development and demonstration. Dr. Schrag reports receiving consultancy or advisory board honoraria from Biogen, Abbvie, Bial, and Neurotechnology; research support from the National Institute of Health Research, Parkinsons UK, and the Economic and Social Research Council and the European Commission; and Royalties from Oxford University Press. Dr. Pringsheim reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Newborn Screening for Spinal Muscular Atrophy in the United States
Click here to read the supplement

Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.
Click here to read the supplement
Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.
Click here to read the supplement
Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.

FDA approves new myasthenia gravis drug
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
Expected spike in acute flaccid myelitis did not occur in 2020
suggested researchers at the Centers for Disease Control and Prevention.
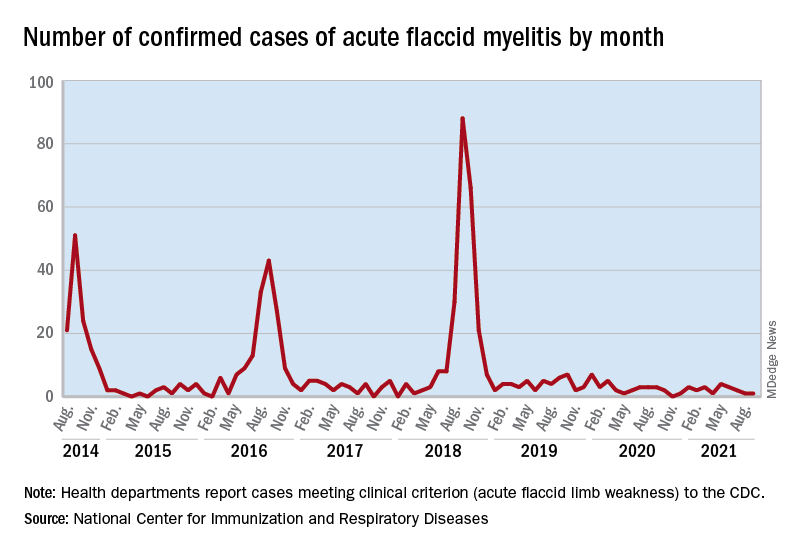
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
FROM MMWR
A safer way to use Botox to treat challenging dystonia type?
, new research suggests.
Oromandibular dystonia causes an involuntary opening of the mouth, which can be disabling and disfiguring. Although injection of the lateral pterygoid muscle with botulinum toxin is the preferred treatment for oromandibular dystonia, a potential complication concerns the maxillary artery, which can run either lateral or medial to the lateral pterygoid muscle.
In a study of 200 Turkish patients, researchers documented significant variations between men and women in the anatomical location of the maxillary artery – and even found lateral versus medial differences on the left and right side in the same individual.
“The results showed that the maxillary artery runs lateral to the muscle in 67% of the Turkish patients,” Rezzak Yilmaz, MD, department of neurology, University of Ankara Medical School, Turkey, reported at the International Congress of Parkinson’s Disease and Movement Disorders.
Given this high rate, there is a high risk for injury “that may result in pain and hematoma” when using preauricular extraoral injections, Dr. Yilmaz and colleagues noted. Instead, they recommend an intraoral injection approach to the lateral pterygoid muscle. “However, this critical anatomical variation is still unrecognized by most clinicians performing [botulinum toxin] injections,” they wrote.
Significant gender differences
The maxillary artery is the largest branch of the external carotid artery.
In the current study, the researchers used magnetic resonance angiography to assess the relevant anatomy in a cohort of 200 individuals (mean age, 56.4 years; 64% women) without a history of facial trauma or movement disorders.
Results showed that the maxillary artery ran lateral to the lateral pterygoid muscle in 67% of the study population.
“This result was also more frequent in females compared with males. Also, there was a considerable variability between the left and the right side in 20% of the participants,” Dr. Yilmaz reported.
Statistically significant gender differences were found for the artery running lateral to the lateral pterygoid muscle on both sides (71.1% in women vs. 58.5% in men; P = .007) and for the artery running lateral to the lateral pterygoid muscle on just the left side (69.8% in women vs. 53.5% in men; P = .02).
In an email exchange, Dr. Yilmaz said if medical personnel are not trained to perform an intraoral approach, “imaging to visualize the path of the maxillary artery before an extraoral/transcutaneous injection can be recommended.”
“If the imaging reveals that the maxillary artery passes lateral to the muscle, then the patient needs to be referred to another center for an intraoral injection,” unless the clinician is trained for an intraoral approach, he added.
Useful education
Commenting on the study, Michele Tagliati, MD, director of the Movement Disorders Program at Cedars-Sinai Medical Center, Los Angeles, said the results were educational. “I didn’t know about all this variability. I was working under the assumption that the artery was medial,” said Dr. Tagliati, who was not involved with the research.
Among his large practice of about 2,000 patients, Dr. Tagliati estimated having five patients for whom he provides this type of injection – and has never encountered a problem with them.
“Maybe all my patients are medial, but now that I’m aware I’ll probably pay more attention,” Dr. Tagliati said. He does not currently perform magnetic resonance angiography before injecting them, “although maybe I should,” he said.
When asked if it is worth the time and expense to perform magnetic resonance angiography on every patient who comes in for lateral pterygoid muscle injections, Dr. Tagliati said that although he has done the injections without problems in his current patients, he may “start obtaining imaging studies to make sure that we’re not taking unnecessary risk” if the maxillary artery is lateral to the lateral pterygoid muscle in new patients.
If there is a risk, he’ll then consider talking with colleagues in oral or facial surgery. Dr. Tagliati added that the number of patients he sees with oromandibular dystonia is rather small, so this extra step would not add a lot of additional imaging.
Overall, Dr. Tagliati noted that the study outcome was significant enough to want to use it for professional education. “I can definitely tell you that I’m going to bring it to the attention of my Fellows. [Every year] I teach one or two Fellows to inject Botox,” he said.
There was no funding for the study. Dr. Yilmaz and Dr. Tagliati have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Oromandibular dystonia causes an involuntary opening of the mouth, which can be disabling and disfiguring. Although injection of the lateral pterygoid muscle with botulinum toxin is the preferred treatment for oromandibular dystonia, a potential complication concerns the maxillary artery, which can run either lateral or medial to the lateral pterygoid muscle.
In a study of 200 Turkish patients, researchers documented significant variations between men and women in the anatomical location of the maxillary artery – and even found lateral versus medial differences on the left and right side in the same individual.
“The results showed that the maxillary artery runs lateral to the muscle in 67% of the Turkish patients,” Rezzak Yilmaz, MD, department of neurology, University of Ankara Medical School, Turkey, reported at the International Congress of Parkinson’s Disease and Movement Disorders.
Given this high rate, there is a high risk for injury “that may result in pain and hematoma” when using preauricular extraoral injections, Dr. Yilmaz and colleagues noted. Instead, they recommend an intraoral injection approach to the lateral pterygoid muscle. “However, this critical anatomical variation is still unrecognized by most clinicians performing [botulinum toxin] injections,” they wrote.
Significant gender differences
The maxillary artery is the largest branch of the external carotid artery.
In the current study, the researchers used magnetic resonance angiography to assess the relevant anatomy in a cohort of 200 individuals (mean age, 56.4 years; 64% women) without a history of facial trauma or movement disorders.
Results showed that the maxillary artery ran lateral to the lateral pterygoid muscle in 67% of the study population.
“This result was also more frequent in females compared with males. Also, there was a considerable variability between the left and the right side in 20% of the participants,” Dr. Yilmaz reported.
Statistically significant gender differences were found for the artery running lateral to the lateral pterygoid muscle on both sides (71.1% in women vs. 58.5% in men; P = .007) and for the artery running lateral to the lateral pterygoid muscle on just the left side (69.8% in women vs. 53.5% in men; P = .02).
In an email exchange, Dr. Yilmaz said if medical personnel are not trained to perform an intraoral approach, “imaging to visualize the path of the maxillary artery before an extraoral/transcutaneous injection can be recommended.”
“If the imaging reveals that the maxillary artery passes lateral to the muscle, then the patient needs to be referred to another center for an intraoral injection,” unless the clinician is trained for an intraoral approach, he added.
Useful education
Commenting on the study, Michele Tagliati, MD, director of the Movement Disorders Program at Cedars-Sinai Medical Center, Los Angeles, said the results were educational. “I didn’t know about all this variability. I was working under the assumption that the artery was medial,” said Dr. Tagliati, who was not involved with the research.
Among his large practice of about 2,000 patients, Dr. Tagliati estimated having five patients for whom he provides this type of injection – and has never encountered a problem with them.
“Maybe all my patients are medial, but now that I’m aware I’ll probably pay more attention,” Dr. Tagliati said. He does not currently perform magnetic resonance angiography before injecting them, “although maybe I should,” he said.
When asked if it is worth the time and expense to perform magnetic resonance angiography on every patient who comes in for lateral pterygoid muscle injections, Dr. Tagliati said that although he has done the injections without problems in his current patients, he may “start obtaining imaging studies to make sure that we’re not taking unnecessary risk” if the maxillary artery is lateral to the lateral pterygoid muscle in new patients.
If there is a risk, he’ll then consider talking with colleagues in oral or facial surgery. Dr. Tagliati added that the number of patients he sees with oromandibular dystonia is rather small, so this extra step would not add a lot of additional imaging.
Overall, Dr. Tagliati noted that the study outcome was significant enough to want to use it for professional education. “I can definitely tell you that I’m going to bring it to the attention of my Fellows. [Every year] I teach one or two Fellows to inject Botox,” he said.
There was no funding for the study. Dr. Yilmaz and Dr. Tagliati have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Oromandibular dystonia causes an involuntary opening of the mouth, which can be disabling and disfiguring. Although injection of the lateral pterygoid muscle with botulinum toxin is the preferred treatment for oromandibular dystonia, a potential complication concerns the maxillary artery, which can run either lateral or medial to the lateral pterygoid muscle.
In a study of 200 Turkish patients, researchers documented significant variations between men and women in the anatomical location of the maxillary artery – and even found lateral versus medial differences on the left and right side in the same individual.
“The results showed that the maxillary artery runs lateral to the muscle in 67% of the Turkish patients,” Rezzak Yilmaz, MD, department of neurology, University of Ankara Medical School, Turkey, reported at the International Congress of Parkinson’s Disease and Movement Disorders.
Given this high rate, there is a high risk for injury “that may result in pain and hematoma” when using preauricular extraoral injections, Dr. Yilmaz and colleagues noted. Instead, they recommend an intraoral injection approach to the lateral pterygoid muscle. “However, this critical anatomical variation is still unrecognized by most clinicians performing [botulinum toxin] injections,” they wrote.
Significant gender differences
The maxillary artery is the largest branch of the external carotid artery.
In the current study, the researchers used magnetic resonance angiography to assess the relevant anatomy in a cohort of 200 individuals (mean age, 56.4 years; 64% women) without a history of facial trauma or movement disorders.
Results showed that the maxillary artery ran lateral to the lateral pterygoid muscle in 67% of the study population.
“This result was also more frequent in females compared with males. Also, there was a considerable variability between the left and the right side in 20% of the participants,” Dr. Yilmaz reported.
Statistically significant gender differences were found for the artery running lateral to the lateral pterygoid muscle on both sides (71.1% in women vs. 58.5% in men; P = .007) and for the artery running lateral to the lateral pterygoid muscle on just the left side (69.8% in women vs. 53.5% in men; P = .02).
In an email exchange, Dr. Yilmaz said if medical personnel are not trained to perform an intraoral approach, “imaging to visualize the path of the maxillary artery before an extraoral/transcutaneous injection can be recommended.”
“If the imaging reveals that the maxillary artery passes lateral to the muscle, then the patient needs to be referred to another center for an intraoral injection,” unless the clinician is trained for an intraoral approach, he added.
Useful education
Commenting on the study, Michele Tagliati, MD, director of the Movement Disorders Program at Cedars-Sinai Medical Center, Los Angeles, said the results were educational. “I didn’t know about all this variability. I was working under the assumption that the artery was medial,” said Dr. Tagliati, who was not involved with the research.
Among his large practice of about 2,000 patients, Dr. Tagliati estimated having five patients for whom he provides this type of injection – and has never encountered a problem with them.
“Maybe all my patients are medial, but now that I’m aware I’ll probably pay more attention,” Dr. Tagliati said. He does not currently perform magnetic resonance angiography before injecting them, “although maybe I should,” he said.
When asked if it is worth the time and expense to perform magnetic resonance angiography on every patient who comes in for lateral pterygoid muscle injections, Dr. Tagliati said that although he has done the injections without problems in his current patients, he may “start obtaining imaging studies to make sure that we’re not taking unnecessary risk” if the maxillary artery is lateral to the lateral pterygoid muscle in new patients.
If there is a risk, he’ll then consider talking with colleagues in oral or facial surgery. Dr. Tagliati added that the number of patients he sees with oromandibular dystonia is rather small, so this extra step would not add a lot of additional imaging.
Overall, Dr. Tagliati noted that the study outcome was significant enough to want to use it for professional education. “I can definitely tell you that I’m going to bring it to the attention of my Fellows. [Every year] I teach one or two Fellows to inject Botox,” he said.
There was no funding for the study. Dr. Yilmaz and Dr. Tagliati have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM MDS VIRTUAL CONGRESS 2021
Investigative botulinum toxin formulation shows prolonged effect
, according to results of a phase 3 clinical trial presented at the virtual International Congress of Parkinson’s Disease and Movement Disorders.

The ASPEN-1 trial evaluated 301 patients with moderate to severe cervical dystonia for up to 36 weeks and found that those receiving two doses of DaxibotulinumtoxinA, known as DAXI, versus placebo improved their scores on the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), said Joseph Jankovic, MD, professor of neurology and director of the Parkinson’s Disease Center and Movement Disorders Clinic at Baylor College of Medicine in Houston.
“Botulinum neurotoxin is clearly the treatment of choice for cervical dystonia,” Dr. Jankovic said in an interview. “While the majority of patients obtain satisfactory benefit from BoNT injections, some experience adverse effects such as neck weakness and difficulty swallowing.” Another limitation of BoNT is that its effects wear off after about 3 months or less and patients have to be re-injected, he said.
“This is why I am quite encouraged by the results of the DAXI study that suggest that this formulation of BoNT (type A) may have a longer response and relatively few side effects,” he said.
Patients in the study were randomized 1:3:3 to placebo, DAXI 125U or DAXI 250U. The average TWSTRS score upon enrollment was 43.3. The placebo group had a mean ± standard error TWSTRS improvement of 4.3 ± 1.8 at 4 or 6 weeks, while the treatment groups had mean ± SE improvements of 12.7 ± 1.3 for 125U and 10.9 ± 1.2 for 250U (P = .0006 vs. placebo). They translate into improvements of 12%, 31%, and 27% for the placebo and low- and high-dose treatment groups, respectively.
“Even though paradoxically it seems the high-dose group did slightly less well than the low-dose group, there was no difference between the two groups,” Dr. Jankovic said in the presentation.
The median duration of benefit was 24 weeks in the low-dose group and 20.3 weeks in the high-dose group.
The treatment groups demonstrated similar benefit compared with placebo in TWSTRS subscales for disease severity, disability, and pain, Dr. Jankovic said. “The majority of the patients had little better, moderately better, or very much better from the botulinum toxin injection with respect to clinical global impression of change and patient global impression of change,” he said.
Likewise, both the Clinician Global Impression of Change (CGIC) and Patient Global Impression of Change (PGIC) demonstrated improvement versus placebo: 77.6% and 76.9% in the 125U and 250U doses versus 45.7% for the former; and 71.2% and 73.1% versus 41.3% for the latter.
Side effects “were remarkably minimal,” Dr. Jankovic said, “but I want to call attention to the low frequency of neck weakness or dysphagia in comparison with other studies of botulinum toxin in cervical dystonia.” The rates of dysphagia were 1.6% and 3.9% in the 125U and 250U treatment groups, respectively. Sixteen of the 255 patients in the treatment groups reported muscular weakness or musculoskeletal pain, and seven had dysphagia.
The rate of dysphagia after injection is noteworthy, said David Charles, MD, professor and vice chair of neurology at Vanderbilt University in Nashville, Tenn., who was not involved in the research. “The one thing we worry about most in people with cervical dystonia are swallowing and choking – dysphagia – and the numbers are very modest: 2 out of 127 in the 125U dose and 5 of 130 in the 250U dose,” he said. “That’s a very low rate of that adverse event.”
The duration of action for both doses is “rather remarkable,” Dr. Charles said. “With the other formulations, my patients are coming back every 12 weeks for treatment; the BoNT helps so much that [these] patients make their appointments every 3 months for as far out as they can,” he said. “This could potentially mean two or three trips a year as opposed to four trips a year.”
The trial was funded by Revance Therapeutics. Dr. Jankovic is an investigator for Revance, and three coauthors are employees of Revance. Dr. Charles is a consultant to the company.
, according to results of a phase 3 clinical trial presented at the virtual International Congress of Parkinson’s Disease and Movement Disorders.
The ASPEN-1 trial evaluated 301 patients with moderate to severe cervical dystonia for up to 36 weeks and found that those receiving two doses of DaxibotulinumtoxinA, known as DAXI, versus placebo improved their scores on the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), said Joseph Jankovic, MD, professor of neurology and director of the Parkinson’s Disease Center and Movement Disorders Clinic at Baylor College of Medicine in Houston.
“Botulinum neurotoxin is clearly the treatment of choice for cervical dystonia,” Dr. Jankovic said in an interview. “While the majority of patients obtain satisfactory benefit from BoNT injections, some experience adverse effects such as neck weakness and difficulty swallowing.” Another limitation of BoNT is that its effects wear off after about 3 months or less and patients have to be re-injected, he said.
“This is why I am quite encouraged by the results of the DAXI study that suggest that this formulation of BoNT (type A) may have a longer response and relatively few side effects,” he said.
Patients in the study were randomized 1:3:3 to placebo, DAXI 125U or DAXI 250U. The average TWSTRS score upon enrollment was 43.3. The placebo group had a mean ± standard error TWSTRS improvement of 4.3 ± 1.8 at 4 or 6 weeks, while the treatment groups had mean ± SE improvements of 12.7 ± 1.3 for 125U and 10.9 ± 1.2 for 250U (P = .0006 vs. placebo). They translate into improvements of 12%, 31%, and 27% for the placebo and low- and high-dose treatment groups, respectively.
“Even though paradoxically it seems the high-dose group did slightly less well than the low-dose group, there was no difference between the two groups,” Dr. Jankovic said in the presentation.
The median duration of benefit was 24 weeks in the low-dose group and 20.3 weeks in the high-dose group.
The treatment groups demonstrated similar benefit compared with placebo in TWSTRS subscales for disease severity, disability, and pain, Dr. Jankovic said. “The majority of the patients had little better, moderately better, or very much better from the botulinum toxin injection with respect to clinical global impression of change and patient global impression of change,” he said.
Likewise, both the Clinician Global Impression of Change (CGIC) and Patient Global Impression of Change (PGIC) demonstrated improvement versus placebo: 77.6% and 76.9% in the 125U and 250U doses versus 45.7% for the former; and 71.2% and 73.1% versus 41.3% for the latter.
Side effects “were remarkably minimal,” Dr. Jankovic said, “but I want to call attention to the low frequency of neck weakness or dysphagia in comparison with other studies of botulinum toxin in cervical dystonia.” The rates of dysphagia were 1.6% and 3.9% in the 125U and 250U treatment groups, respectively. Sixteen of the 255 patients in the treatment groups reported muscular weakness or musculoskeletal pain, and seven had dysphagia.
The rate of dysphagia after injection is noteworthy, said David Charles, MD, professor and vice chair of neurology at Vanderbilt University in Nashville, Tenn., who was not involved in the research. “The one thing we worry about most in people with cervical dystonia are swallowing and choking – dysphagia – and the numbers are very modest: 2 out of 127 in the 125U dose and 5 of 130 in the 250U dose,” he said. “That’s a very low rate of that adverse event.”
The duration of action for both doses is “rather remarkable,” Dr. Charles said. “With the other formulations, my patients are coming back every 12 weeks for treatment; the BoNT helps so much that [these] patients make their appointments every 3 months for as far out as they can,” he said. “This could potentially mean two or three trips a year as opposed to four trips a year.”
The trial was funded by Revance Therapeutics. Dr. Jankovic is an investigator for Revance, and three coauthors are employees of Revance. Dr. Charles is a consultant to the company.
, according to results of a phase 3 clinical trial presented at the virtual International Congress of Parkinson’s Disease and Movement Disorders.
The ASPEN-1 trial evaluated 301 patients with moderate to severe cervical dystonia for up to 36 weeks and found that those receiving two doses of DaxibotulinumtoxinA, known as DAXI, versus placebo improved their scores on the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), said Joseph Jankovic, MD, professor of neurology and director of the Parkinson’s Disease Center and Movement Disorders Clinic at Baylor College of Medicine in Houston.
“Botulinum neurotoxin is clearly the treatment of choice for cervical dystonia,” Dr. Jankovic said in an interview. “While the majority of patients obtain satisfactory benefit from BoNT injections, some experience adverse effects such as neck weakness and difficulty swallowing.” Another limitation of BoNT is that its effects wear off after about 3 months or less and patients have to be re-injected, he said.
“This is why I am quite encouraged by the results of the DAXI study that suggest that this formulation of BoNT (type A) may have a longer response and relatively few side effects,” he said.
Patients in the study were randomized 1:3:3 to placebo, DAXI 125U or DAXI 250U. The average TWSTRS score upon enrollment was 43.3. The placebo group had a mean ± standard error TWSTRS improvement of 4.3 ± 1.8 at 4 or 6 weeks, while the treatment groups had mean ± SE improvements of 12.7 ± 1.3 for 125U and 10.9 ± 1.2 for 250U (P = .0006 vs. placebo). They translate into improvements of 12%, 31%, and 27% for the placebo and low- and high-dose treatment groups, respectively.
“Even though paradoxically it seems the high-dose group did slightly less well than the low-dose group, there was no difference between the two groups,” Dr. Jankovic said in the presentation.
The median duration of benefit was 24 weeks in the low-dose group and 20.3 weeks in the high-dose group.
The treatment groups demonstrated similar benefit compared with placebo in TWSTRS subscales for disease severity, disability, and pain, Dr. Jankovic said. “The majority of the patients had little better, moderately better, or very much better from the botulinum toxin injection with respect to clinical global impression of change and patient global impression of change,” he said.
Likewise, both the Clinician Global Impression of Change (CGIC) and Patient Global Impression of Change (PGIC) demonstrated improvement versus placebo: 77.6% and 76.9% in the 125U and 250U doses versus 45.7% for the former; and 71.2% and 73.1% versus 41.3% for the latter.
Side effects “were remarkably minimal,” Dr. Jankovic said, “but I want to call attention to the low frequency of neck weakness or dysphagia in comparison with other studies of botulinum toxin in cervical dystonia.” The rates of dysphagia were 1.6% and 3.9% in the 125U and 250U treatment groups, respectively. Sixteen of the 255 patients in the treatment groups reported muscular weakness or musculoskeletal pain, and seven had dysphagia.
The rate of dysphagia after injection is noteworthy, said David Charles, MD, professor and vice chair of neurology at Vanderbilt University in Nashville, Tenn., who was not involved in the research. “The one thing we worry about most in people with cervical dystonia are swallowing and choking – dysphagia – and the numbers are very modest: 2 out of 127 in the 125U dose and 5 of 130 in the 250U dose,” he said. “That’s a very low rate of that adverse event.”
The duration of action for both doses is “rather remarkable,” Dr. Charles said. “With the other formulations, my patients are coming back every 12 weeks for treatment; the BoNT helps so much that [these] patients make their appointments every 3 months for as far out as they can,” he said. “This could potentially mean two or three trips a year as opposed to four trips a year.”
The trial was funded by Revance Therapeutics. Dr. Jankovic is an investigator for Revance, and three coauthors are employees of Revance. Dr. Charles is a consultant to the company.
FROM MDS VIRTUAL CONGRESS 2021
Synthetic triglyceride shows potential in Huntington’s disease
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.

Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.
As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.
Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.
As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
, according to data presented at the International Congress of Parkinson’s Disease and Movement Disorders.
Reporting results of TRIHEP3 and an extension study, Fanny Mochel, MD, PhD, of Sorbonne University in Paris and the Paris Brain Institute, said in an interview that her group is the only one investigating triheptanoin to target caudate atrophy in Huntington’s disease. The Food and Drug Administration last year approved triheptanoin for the treatment of long-chain fatty acid oxidation disorders.
“The main findings are two observations: that patients were clinically stable based on their gradation of total motor score (TMS) on UHDRS (Unified Huntington’s Disease Rating Scale) after 1 year,” Dr. Mochel said in an interview. “The other is that we observed a reduction of the caudate atrophy progression that we usually see over 1 year by about 50%.”
TRIHEP3 randomized 100 patients with early-stage Huntington’s disease to triheptanoin 1g/kg daily and placebo. It followed on previous research in which the group used 31-phosphorus brain MR spectroscopy to demonstrate triheptanoin restored a normal brain energetic profile in patients with Huntington’s disease. TRIHEP3 was a 6-month randomized controlled trial at two centers, followed by a 6-month open-label phase. After that, 42 patients opted to participate in the 1-year extension study.
TRIHEP3 found no difference in caudate boundary shift integral (cBSI) at 6 months – the primary endpoint. But in the extension study, TMS tended to stabilize in patients treated for 1 year (0.6 ± 5.1), compared with those treated for 6 months (2.5 ± 4.5, P = .072).
Using a placebo control group from an external study of patients with Huntington’s disease with what Dr. Mochel described as “identical clinical characteristics,” she said the research confirmed TMS clinical stability in treated patients at 1 year (2.6 ± 4.6 vs. 0.6 ± 5.1, P = .057) and found significantly lower caudate atrophy (–3% vs. –6.7%, compared with baseline, P < .001).
Dr. Mochel also noted that Diffusion Tensor Imaging and Fixed-based analyses (FBA) showed fewer alterations in fiber metrics at 24 months in patients treated from baseline. FBA also showed improved fiber trophicity at 24 months in both groups.
‘The first good news’
Dr. Mochel noted that the Huntington’s disease community had been shaken in the spring by the failure of three trials of gene-targeting therapies for Huntington’s disease. Roche halted a phase 3 study of its antisense oligonucleotide (ASO) tominersen, and Wave Life Sciences scuttled two ASO programs in phase 1/2 trials.
“Triheptanoin is not going to cure Huntington’s disease; it’s a disease with many components, but it does work on the energy aspects and that seems to stabilize patients over the time of observation,” Dr. Mochel said. “That’s the first good news.”
She also noted that side effects were mainly gastrointestinal in nature, and they typically resolved with dietary management.
As a target in Huntington disease, the caudate nucleus is highly desirable, and caudate atrophy has been shown to occur even before the onset of motor symptoms, said N. Ahmad Aziz, MD, PhD, a neurologist and epidemiologist at the German Center for Neurodegenerative Diseases at the University of Bonn (Germany). “In this light, the findings of the trial conducted by Dr. Mochel and colleagues, which suggest that triheptanoin intake may slow down the rate of caudate atrophy in patients with early-stage Huntington’s disease, are highly promising,” Dr. Aziz said in an interview.
However, he noted that the improvement in caudate atrophy was only a secondary endpoint in the extension study. “Nevertheless, given triheptanoin’s biologically plausible mechanism of action – i.e., provision of substrates to the Krebs cycle and at least partial restoration of the well-documented defective mitochondrial function in Huntington’s disease – combined with its apparently relatively mild side-effect profile and good tolerability, I think that the preliminary findings of this trial are very promising and justify a larger phase 3 trial,” Dr. Aziz said.
Dr. Mochel said that the findings are prompting the investigators to consider just that.
Dr. Mochel has received consulting fees from and conducted investigator‐sponsored studies supported by Ultragenyx Pharmaceuticals. Dr. Aziz has no relevant financial relationships to disclose.
FROM MDS VIRTUAL CONGRESS 2021
Potential first-in-class, targeted therapy for myasthenia gravis
(gMG), new research suggests. Results from the phase 3, randomized, placebo-controlled ADAPT trial showed that reduction in disease burden and improvement in strength and quality of life in patients with gMG were consistent across four MG-specific scales for those receiving the novel treatment. In addition, these benefits were observed early and were reproducible and durable, the researchers noted.
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work, and the side effect profile is much like placebo,” said principal investigator James Howard, Jr., MD, Distinguished Professor of Neuromuscular Disease, department of neurology, University of North Carolina at Chapel Hill.
The ADAPT results are “important for the MG community, and I am hopeful efgartigimod will provide a first-in-class targeted therapy that can be dosed in an individual way for people living with this chronic autoimmune disease,” Dr. Howard added in a news release.
The findings were published online June 17 in Lancet Neurology.
Targeted molecular therapy
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have immunoglobulin G (IgG) antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an investigational antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
The ADAPT trial was conducted at 56 neuromuscular academic and community centers in 15 countries in North America, Europe, and Japan. The study included 167 adults with gMG, regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis Activities of Daily Living (MG-ADL) score of at least 5 (greater than 50% non-ocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
ADAPT was designed to allow an individualized treatment approach with an initial treatment cycle followed by a variable number of subsequent treatment cycles, the investigators noted.
Primary endpoint met
The primary efficacy endpoint was number of acetylcholine receptor antibody-positive (AChR-Ab+) patients who achieved a clinically meaningful response on the MG-ADL score. This was defined as at least a 2-point improvement from baseline for 4 or more consecutive weeks. Forty-four (68%) of 65 AChR-Ab+ patients treated with efgartigimod met this endpoint versus 19 (30%) of 64 patients treated with placebo (odds ratio, 4.95; 95% confidence interval, 2.21-11.53; P < .0001).
Many of the patients treated with efgartigimod showed improvement “beyond the clinically meaningful threshold, achieving up to 9-point reductions in MG-ADL,” the investigators reported. In addition, 40% of the efgartigimod group attained an MG-ADL score of 0 or 1 (minimal symptom expression) in cycle 1 versus 11% of the placebo group (P < .0001).
Nearly two-thirds (63%) of AChR-Ab+ patients responded to the first cycle of efgartigimod, and most of these patients (83%) responded to treatment within the first 2 weeks. Among the AChR-Ab+ participants who responded to efgartigimod in cycle 1, the duration of responder status was 6 to 7 weeks in 32% of patients, 8 to 11 weeks in 23% of patients, and 12 weeks or more in 34% of patients.
Safety profile
“Some patients never required retreatment over the 26-week period that they were under observation,” Dr. Howard said. “Patients want to be individuals. They don’t want to be assigned to a regimented therapy, and I think these results show that this therapy can be tailored to the individual patient, rather than simply giving it to them in a cookbook fashion,” he added.
The safety profile of efgartigimod was comparable to placebo. Most adverse events were mild or moderate in severity. The most commonly reported adverse events were headache, nasopharyngitis, nausea, diarrhea, upper respiratory tract infection, and urinary tract infection.
Four (5%) efgartigimod-treated patients had a serious adverse event, which included thrombocytosis, rectal adenocarcinoma, worsening MG, and depression.
The novel agent is currently under review with the U.S. Food and Drug Administration for the treatment of gMG, with a Prescription Drug User Fee Act target action date of Dec. 17. If approved, it would become the first FDA-approved FcRn antagonist.
Expanding therapeutic landscape
In a linked commentary, Shigeaki Suzuki, MD, PhD, department of neurology, Keio University School of Medicine, Tokyo, noted that the therapeutic landscape for patients with MG is “expanding year by year,” with several additional complement inhibitors and FcRn antagonists now in phase 3 testing.
“Biological drugs should be preferentially used as the treatment for patients with refractory myasthenia gravis, although the definition of refractory myasthenia gravis is different depending on the criteria used,” Dr. Suzuki wrote.
He noted that when “cost-effectiveness is not taken into account, targeted molecular therapy might be used widely” in patients with MG.
“Risks of myasthenic exacerbation and crises should be substantially decreased, particularly in patients with refractory myasthenia gravis,” Dr. Suzuki added.
The ADAPT study was supported by argenx. Dr. Howard has reported receiving research support from argenx, Alexion Pharmaceuticals, the Centers for Disease Control and Prevention, the Muscular Dystrophy Association, the National Institutes of Health, Patient-Centered Outcomes Research Institute, and Ra Pharmaceuticals; honoraria from argenx, Alexion, Immunovant, Ra, Regeneron Pharmaceuticals, and Viela Bio; and nonfinancial support from argenx, Alexion, Ra, and Toleranzia. Disclosures for the other investigators are listed in the original article. Dr. Suzuki has reported relationships with Alexion Pharmaceuticals, Japan Blood Products Organization, and Asahi Kasei Medical.
A version of this article first appeared on Medscape.com.
(gMG), new research suggests. Results from the phase 3, randomized, placebo-controlled ADAPT trial showed that reduction in disease burden and improvement in strength and quality of life in patients with gMG were consistent across four MG-specific scales for those receiving the novel treatment. In addition, these benefits were observed early and were reproducible and durable, the researchers noted.
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work, and the side effect profile is much like placebo,” said principal investigator James Howard, Jr., MD, Distinguished Professor of Neuromuscular Disease, department of neurology, University of North Carolina at Chapel Hill.
The ADAPT results are “important for the MG community, and I am hopeful efgartigimod will provide a first-in-class targeted therapy that can be dosed in an individual way for people living with this chronic autoimmune disease,” Dr. Howard added in a news release.
The findings were published online June 17 in Lancet Neurology.
Targeted molecular therapy
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have immunoglobulin G (IgG) antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an investigational antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
The ADAPT trial was conducted at 56 neuromuscular academic and community centers in 15 countries in North America, Europe, and Japan. The study included 167 adults with gMG, regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis Activities of Daily Living (MG-ADL) score of at least 5 (greater than 50% non-ocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
ADAPT was designed to allow an individualized treatment approach with an initial treatment cycle followed by a variable number of subsequent treatment cycles, the investigators noted.
Primary endpoint met
The primary efficacy endpoint was number of acetylcholine receptor antibody-positive (AChR-Ab+) patients who achieved a clinically meaningful response on the MG-ADL score. This was defined as at least a 2-point improvement from baseline for 4 or more consecutive weeks. Forty-four (68%) of 65 AChR-Ab+ patients treated with efgartigimod met this endpoint versus 19 (30%) of 64 patients treated with placebo (odds ratio, 4.95; 95% confidence interval, 2.21-11.53; P < .0001).
Many of the patients treated with efgartigimod showed improvement “beyond the clinically meaningful threshold, achieving up to 9-point reductions in MG-ADL,” the investigators reported. In addition, 40% of the efgartigimod group attained an MG-ADL score of 0 or 1 (minimal symptom expression) in cycle 1 versus 11% of the placebo group (P < .0001).
Nearly two-thirds (63%) of AChR-Ab+ patients responded to the first cycle of efgartigimod, and most of these patients (83%) responded to treatment within the first 2 weeks. Among the AChR-Ab+ participants who responded to efgartigimod in cycle 1, the duration of responder status was 6 to 7 weeks in 32% of patients, 8 to 11 weeks in 23% of patients, and 12 weeks or more in 34% of patients.
Safety profile
“Some patients never required retreatment over the 26-week period that they were under observation,” Dr. Howard said. “Patients want to be individuals. They don’t want to be assigned to a regimented therapy, and I think these results show that this therapy can be tailored to the individual patient, rather than simply giving it to them in a cookbook fashion,” he added.
The safety profile of efgartigimod was comparable to placebo. Most adverse events were mild or moderate in severity. The most commonly reported adverse events were headache, nasopharyngitis, nausea, diarrhea, upper respiratory tract infection, and urinary tract infection.
Four (5%) efgartigimod-treated patients had a serious adverse event, which included thrombocytosis, rectal adenocarcinoma, worsening MG, and depression.
The novel agent is currently under review with the U.S. Food and Drug Administration for the treatment of gMG, with a Prescription Drug User Fee Act target action date of Dec. 17. If approved, it would become the first FDA-approved FcRn antagonist.
Expanding therapeutic landscape
In a linked commentary, Shigeaki Suzuki, MD, PhD, department of neurology, Keio University School of Medicine, Tokyo, noted that the therapeutic landscape for patients with MG is “expanding year by year,” with several additional complement inhibitors and FcRn antagonists now in phase 3 testing.
“Biological drugs should be preferentially used as the treatment for patients with refractory myasthenia gravis, although the definition of refractory myasthenia gravis is different depending on the criteria used,” Dr. Suzuki wrote.
He noted that when “cost-effectiveness is not taken into account, targeted molecular therapy might be used widely” in patients with MG.
“Risks of myasthenic exacerbation and crises should be substantially decreased, particularly in patients with refractory myasthenia gravis,” Dr. Suzuki added.
The ADAPT study was supported by argenx. Dr. Howard has reported receiving research support from argenx, Alexion Pharmaceuticals, the Centers for Disease Control and Prevention, the Muscular Dystrophy Association, the National Institutes of Health, Patient-Centered Outcomes Research Institute, and Ra Pharmaceuticals; honoraria from argenx, Alexion, Immunovant, Ra, Regeneron Pharmaceuticals, and Viela Bio; and nonfinancial support from argenx, Alexion, Ra, and Toleranzia. Disclosures for the other investigators are listed in the original article. Dr. Suzuki has reported relationships with Alexion Pharmaceuticals, Japan Blood Products Organization, and Asahi Kasei Medical.
A version of this article first appeared on Medscape.com.
(gMG), new research suggests. Results from the phase 3, randomized, placebo-controlled ADAPT trial showed that reduction in disease burden and improvement in strength and quality of life in patients with gMG were consistent across four MG-specific scales for those receiving the novel treatment. In addition, these benefits were observed early and were reproducible and durable, the researchers noted.
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work, and the side effect profile is much like placebo,” said principal investigator James Howard, Jr., MD, Distinguished Professor of Neuromuscular Disease, department of neurology, University of North Carolina at Chapel Hill.
The ADAPT results are “important for the MG community, and I am hopeful efgartigimod will provide a first-in-class targeted therapy that can be dosed in an individual way for people living with this chronic autoimmune disease,” Dr. Howard added in a news release.
The findings were published online June 17 in Lancet Neurology.
Targeted molecular therapy
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have immunoglobulin G (IgG) antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an investigational antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
The ADAPT trial was conducted at 56 neuromuscular academic and community centers in 15 countries in North America, Europe, and Japan. The study included 167 adults with gMG, regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis Activities of Daily Living (MG-ADL) score of at least 5 (greater than 50% non-ocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
ADAPT was designed to allow an individualized treatment approach with an initial treatment cycle followed by a variable number of subsequent treatment cycles, the investigators noted.
Primary endpoint met
The primary efficacy endpoint was number of acetylcholine receptor antibody-positive (AChR-Ab+) patients who achieved a clinically meaningful response on the MG-ADL score. This was defined as at least a 2-point improvement from baseline for 4 or more consecutive weeks. Forty-four (68%) of 65 AChR-Ab+ patients treated with efgartigimod met this endpoint versus 19 (30%) of 64 patients treated with placebo (odds ratio, 4.95; 95% confidence interval, 2.21-11.53; P < .0001).
Many of the patients treated with efgartigimod showed improvement “beyond the clinically meaningful threshold, achieving up to 9-point reductions in MG-ADL,” the investigators reported. In addition, 40% of the efgartigimod group attained an MG-ADL score of 0 or 1 (minimal symptom expression) in cycle 1 versus 11% of the placebo group (P < .0001).
Nearly two-thirds (63%) of AChR-Ab+ patients responded to the first cycle of efgartigimod, and most of these patients (83%) responded to treatment within the first 2 weeks. Among the AChR-Ab+ participants who responded to efgartigimod in cycle 1, the duration of responder status was 6 to 7 weeks in 32% of patients, 8 to 11 weeks in 23% of patients, and 12 weeks or more in 34% of patients.
Safety profile
“Some patients never required retreatment over the 26-week period that they were under observation,” Dr. Howard said. “Patients want to be individuals. They don’t want to be assigned to a regimented therapy, and I think these results show that this therapy can be tailored to the individual patient, rather than simply giving it to them in a cookbook fashion,” he added.
The safety profile of efgartigimod was comparable to placebo. Most adverse events were mild or moderate in severity. The most commonly reported adverse events were headache, nasopharyngitis, nausea, diarrhea, upper respiratory tract infection, and urinary tract infection.
Four (5%) efgartigimod-treated patients had a serious adverse event, which included thrombocytosis, rectal adenocarcinoma, worsening MG, and depression.
The novel agent is currently under review with the U.S. Food and Drug Administration for the treatment of gMG, with a Prescription Drug User Fee Act target action date of Dec. 17. If approved, it would become the first FDA-approved FcRn antagonist.
Expanding therapeutic landscape
In a linked commentary, Shigeaki Suzuki, MD, PhD, department of neurology, Keio University School of Medicine, Tokyo, noted that the therapeutic landscape for patients with MG is “expanding year by year,” with several additional complement inhibitors and FcRn antagonists now in phase 3 testing.
“Biological drugs should be preferentially used as the treatment for patients with refractory myasthenia gravis, although the definition of refractory myasthenia gravis is different depending on the criteria used,” Dr. Suzuki wrote.
He noted that when “cost-effectiveness is not taken into account, targeted molecular therapy might be used widely” in patients with MG.
“Risks of myasthenic exacerbation and crises should be substantially decreased, particularly in patients with refractory myasthenia gravis,” Dr. Suzuki added.
The ADAPT study was supported by argenx. Dr. Howard has reported receiving research support from argenx, Alexion Pharmaceuticals, the Centers for Disease Control and Prevention, the Muscular Dystrophy Association, the National Institutes of Health, Patient-Centered Outcomes Research Institute, and Ra Pharmaceuticals; honoraria from argenx, Alexion, Immunovant, Ra, Regeneron Pharmaceuticals, and Viela Bio; and nonfinancial support from argenx, Alexion, Ra, and Toleranzia. Disclosures for the other investigators are listed in the original article. Dr. Suzuki has reported relationships with Alexion Pharmaceuticals, Japan Blood Products Organization, and Asahi Kasei Medical.
A version of this article first appeared on Medscape.com.
From Lancet Neurology