User login
Antibodies Part 3: In whose corner is genome editing’s best cut-man sitting?
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.
In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
vinya@mskcc.org
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.
In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
vinya@mskcc.org
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
We are in the midst of a revolution in genome editing. Science now exists in “AC,” or after CRISPR. Able to speedily and efficiently make genomic cuts with surgical precision, CRISPR/Cas9 is used almost ubiquitously now in the scientific community to study and alter DNA across fields ranging from medicine to agriculture to zoology. The possibilities of the biological and therapeutic implications are seemingly endless, as are the important ethical implications of their impact. Likely because of the latter, CRISPR technology has made its way from publications like Science and Nature into the lay public domains of Newsweek and NBC News.
In fact, CRISPR technology made its way into one of my favorite podcasts, WNYC’s “Radio Lab” in June 20151. The episode was entitled “Antibodies Part 1,” perhaps assuming that other technologies would also be discussed later although that has never happened. Actually, in an update early this year, the podcast jokingly addressed never moving on to “Part 2,” then followed with an update on how far CRISPR technology has progressed. Putting aside the technological advances and the early clinical applications, as well as the immense ethical considerations, CRISPR technology faces a new controversy, not one from a white coat but rather from a black robe.
In her CommonHealth blog2, Carey Goldberg of WBUR Boston Public Radio compared the case with the bout between undefeated Muhammad Ali and undefeated Joe Frazier at New York’s Madison Square Garden. Both men had legitimate claims to the title of World Heavyweight Champion. What transpired is now known as the “Fight of the Century.”
The analogy is apt. Boxing is about speed and control. Ali dominated the first three rounds with his jab, a punch that is both offensive with its attack and defensive in keeping one’s opponent at a distance. Dr. Doudna and her collaborator Emmanuelle Charpentier, PhD, published their work first (Science. 2012 Aug 17;337[6096]:816-21)3. UC Berkeley filed their patent first in May 2012.
Boxing is about timing and opportunity. Under the barrage of Ali’s jabs, Frazier found an inside position and caught Ali with a left hook. Dr. Zhang’s work followed closely after but had previously applied the technology in murine and human cells (Science. 2013 Feb 15; 339[6121]:819-23)4. The Broad Institute used this key difference to apply for its own patents under expedited review, which were granted in April 2014.
Boxing is about a punch and a counterpunch. Though fatigued, Ali continued to connect with combination punches. Frazier’s left hook pummeled Ali’s jaw. UC Berkeley filed an interference motion to invalidate the Broad Institute patent claim on the basis that the extension to eukaryotic cells was “obvious” based on the published work by Dr. Doudna’s group. In February, USPTO ruled that the Broad patent application may proceed, citing “patentably distinct subject matter.” Initial reports had indicated that Berkeley may appeal the decision, but no official filings have been made public.
Like the Fight of the Century, this case may go the distance and be decided by the judges. In a unanimous decision, Joe Frazier won the first of three epic bouts. The final scorecard is not known in the patent disputes; on March 28, the European Patent Office announced it will grant the patent application on behalf of Dr. Doudna and Dr. Charpentier.
Much like a prizefight, Wall Street has also been taking bets on who will prevail, with CRISPR-based biotech backing both sides mirroring the mid-bout odds, just as Ali dominated early with the jab, until Frazier evened the match with a left hook to the jaw. Ali fell to his knee on the canvas in the 11th round; will the European Patent Office decision prove to be a slip or a decisive knockdown? With so much at stake, the only assurance is that, as with the Ali-Frazier bout, there is likely more fighting to be done.
References:
1. Radio Lab
2. CommonHealth blog
3. Jinek M, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
4. Le Cong et al., Multiplex genome engineering using CRISPR/Cas Systems. Science. 2013 Feb 15;339(6121):819-23. Multiplex genome engineering using CRISPR/Cas Systems.
Science. 2017 Feb 15: Round one of CRISPR patent legal battle goes to the Broad Institute.
StreetInsider.com: CRISPR Therapeutics (CRSP) says EPO to grant CRISPR/Cas gene editing patent.
vinya@mskcc.org
On Twitter @thedoctorisvin
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
Researchers find ‘feedback loop’ key to reducing high blood pressure
Research with mice and rats in Germany may have found a new way to treat high blood pressure.
Using epifluorescence intravital video microscopy imaging, researchers examined mice to whom they had given angiotensin II hormones to induce arterial hypertension.
They determined that the mice with low levels of thrombin-driven factor XI (FXI) – either naturally or inhibited by antisense oligonucleotides – had healthier endothelium.
“Specificity of the effects of FXI depletion was confirmed by continuous in vivo supplementation with human FXI,” wrote Sabine Kossmann, PhD, of the Center for Thrombosis and Hemostasis, University Medical Center, Mainz (Germany), and her coauthors (Sci Transl Med. 2017 Feb 1. doi: 10.1126/scitranslmed.aah4923).
Targeting the “feedback loop” between the FXI and a receptor that helps thrombin propagate on platelets reduced both vascular inflammation and blood pressure.
“Our findings suggest that inhibiting the ... thrombin-FXI–amplifying loop may provide added cardiovascular benefits that are synergistic with those of established platelet inhibitors,” the authors wrote.
This work was supported by grants from the Stiftung Pathobiochemie und Molekulare Diagnostik and the Federal Ministry of Education and Research. The authors disclosed funding and grants from the German Research Society, the European Research Council, NIH, and other sources. One of the researchers is inventor of five patents related to the FXI inhibitor and equity holder in Aronora, and may have financial interest in the findings of the research.
Research with mice and rats in Germany may have found a new way to treat high blood pressure.
Using epifluorescence intravital video microscopy imaging, researchers examined mice to whom they had given angiotensin II hormones to induce arterial hypertension.
They determined that the mice with low levels of thrombin-driven factor XI (FXI) – either naturally or inhibited by antisense oligonucleotides – had healthier endothelium.
“Specificity of the effects of FXI depletion was confirmed by continuous in vivo supplementation with human FXI,” wrote Sabine Kossmann, PhD, of the Center for Thrombosis and Hemostasis, University Medical Center, Mainz (Germany), and her coauthors (Sci Transl Med. 2017 Feb 1. doi: 10.1126/scitranslmed.aah4923).
Targeting the “feedback loop” between the FXI and a receptor that helps thrombin propagate on platelets reduced both vascular inflammation and blood pressure.
“Our findings suggest that inhibiting the ... thrombin-FXI–amplifying loop may provide added cardiovascular benefits that are synergistic with those of established platelet inhibitors,” the authors wrote.
This work was supported by grants from the Stiftung Pathobiochemie und Molekulare Diagnostik and the Federal Ministry of Education and Research. The authors disclosed funding and grants from the German Research Society, the European Research Council, NIH, and other sources. One of the researchers is inventor of five patents related to the FXI inhibitor and equity holder in Aronora, and may have financial interest in the findings of the research.
Research with mice and rats in Germany may have found a new way to treat high blood pressure.
Using epifluorescence intravital video microscopy imaging, researchers examined mice to whom they had given angiotensin II hormones to induce arterial hypertension.
They determined that the mice with low levels of thrombin-driven factor XI (FXI) – either naturally or inhibited by antisense oligonucleotides – had healthier endothelium.
“Specificity of the effects of FXI depletion was confirmed by continuous in vivo supplementation with human FXI,” wrote Sabine Kossmann, PhD, of the Center for Thrombosis and Hemostasis, University Medical Center, Mainz (Germany), and her coauthors (Sci Transl Med. 2017 Feb 1. doi: 10.1126/scitranslmed.aah4923).
Targeting the “feedback loop” between the FXI and a receptor that helps thrombin propagate on platelets reduced both vascular inflammation and blood pressure.
“Our findings suggest that inhibiting the ... thrombin-FXI–amplifying loop may provide added cardiovascular benefits that are synergistic with those of established platelet inhibitors,” the authors wrote.
This work was supported by grants from the Stiftung Pathobiochemie und Molekulare Diagnostik and the Federal Ministry of Education and Research. The authors disclosed funding and grants from the German Research Society, the European Research Council, NIH, and other sources. One of the researchers is inventor of five patents related to the FXI inhibitor and equity holder in Aronora, and may have financial interest in the findings of the research.
FROM SCIENCE TRANSLATIONAL MEDICINE
FOUND IN TRANSLATION Minimal nomenclature and maximum sensitivity complicate MRD measures
In hematologic malignancies, there is a deep and direct connection between each individual patient, that patient’s symptoms, the visible cells that cause the disease, and the direct measurements and assessments of those cells. The totality of these factors helps to determine the diagnosis and treatment plan. As a butterfly needle often is sufficient for obtaining a diagnostic tumor biopsy, it is not surprising that these same diagnostic techniques are now standardly being used to monitor disease response.
The techniques differ in their limits of detection, however. With sequencing depths able to reliably detect variant allele frequencies of less than 10%, even when patients’ overt leukemia may no longer be detectable, clinicians may be left to ponder what to do with persistent “preleukemic” or “rising clones.”1-3
These patients, now more appropriately stratified for risk of recurrence, are in desperate need of better care algorithms. Standard MRD assessment by flow cytometric analysis is able to detect less than 1 x 10-4 cells. While it can be applied to most patients, its sensitivity will likely be surpassed by new and emerging genomic assays. Real time quantitative polymerase chain reaction (RT-qPCR) and next generation sequencing (NGS) require a leukemia-specific abnormality but have the potential for far greater sensitivity with deeper sequencing techniques.
Long-term follow up data in acute promyelocytic leukemia (APL) provides the illustrative example where morphologic remission is not durable in the setting of a persistent PML-RARa transcript and therapeutic goals for PCR negativity irrespective of morphology are standard. Pathologic fusion proteins are ideal for marker-driven therapy, but are found in only about 50% of patients, mainly those with APL and Philadelphia chromosome-positive leukemias.
With driver mutations identified in the majority of patients, we can be hopeful that NGS negativity may be a useful clinical endpoint. In work presented at ASH 2016 by Bartlomiej M Getta, MBBS, of Memorial Sloan Kettering Cancer Center, New York, and his colleagues, patients with concordant MRD positivity by flow cytometry and NGS had inferior outcomes, even after allogeneic transplant, compared to patients with MRD positivity on one assay but not both.5 Nonetheless, caution should be taken in early adoption of NGS as a independent marker of MRD status for two main reasons: 1) False positives and lack of standardization make current interpretation difficult. 2) The presence of “preleukemic” clones remains enigmatic – and no matter the nomenclature used, can a DNMT3A or IDH-mutant clone really be deemed “clonal hematopoiesis of indeterminate potential” when a patient has already had clonal transformation?
Conversely, not all patients reported in the work by Klco2 and Getta ultimately relapse. Thus, while it would be preferred to clear all mutant clones, as a therapeutic goal this likely would subject many patients to unnecessary toxicity. One half of the patients reported by Getta were disease free at a year with concordant flow and NGS positive MRD while patients with NGS positivity alone had outcomes equivalent to those of MRD-negative patients, highlighting that certain persistent clones in NGS-only, MRD-positive patients might be amenable to immunotherapy, either with checkpoint inhibitors or allogeneic transplant. Insight into which clones remain quiescent and which are more sinister will require more investigation, but there does seem to be an additive role to NGS-positivity, whereby all MRD is not created equal and the precision and clinical utility of MRD status will likely take on nuanced nomenclature.
References
1. Jan, M. et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine 4, 149ra118, doi: 10.1126/scitranslmed.3004315 (2012).
2. Klco, J. M. et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015;314:811-22. doi: 10.1001/jama.2015.9643.
3. Wong, T. N. et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016;127:893-7. doi: 10.1182/blood-2015-10-677021 (2016).
4. Lane, A. A. et al. Results from Ongoing Phase II Trial of SL-401 As Consolidation Therapy in Patients with Acute Myeloid Leukemia (AML) in Remission with High Relapse Risk Including Minimal Residual Disease (MRD), Abstract 215, ASH 2016.
5. Getta, B. M. et al. Multicolor Flow Cytometry and Multi-Gene Next Generation Sequencing Are Complimentary and Highly Predictive for Relapse in Acute Myeloid Leukemia Following Allogeneic Hematopoietic Stem Cell Transplant, Abstract 834, ASH 2016.
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, New York, where he is a clinical instructor, on the staff of the leukemia service, and a clinical researcher in The Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
In hematologic malignancies, there is a deep and direct connection between each individual patient, that patient’s symptoms, the visible cells that cause the disease, and the direct measurements and assessments of those cells. The totality of these factors helps to determine the diagnosis and treatment plan. As a butterfly needle often is sufficient for obtaining a diagnostic tumor biopsy, it is not surprising that these same diagnostic techniques are now standardly being used to monitor disease response.
The techniques differ in their limits of detection, however. With sequencing depths able to reliably detect variant allele frequencies of less than 10%, even when patients’ overt leukemia may no longer be detectable, clinicians may be left to ponder what to do with persistent “preleukemic” or “rising clones.”1-3
These patients, now more appropriately stratified for risk of recurrence, are in desperate need of better care algorithms. Standard MRD assessment by flow cytometric analysis is able to detect less than 1 x 10-4 cells. While it can be applied to most patients, its sensitivity will likely be surpassed by new and emerging genomic assays. Real time quantitative polymerase chain reaction (RT-qPCR) and next generation sequencing (NGS) require a leukemia-specific abnormality but have the potential for far greater sensitivity with deeper sequencing techniques.
Long-term follow up data in acute promyelocytic leukemia (APL) provides the illustrative example where morphologic remission is not durable in the setting of a persistent PML-RARa transcript and therapeutic goals for PCR negativity irrespective of morphology are standard. Pathologic fusion proteins are ideal for marker-driven therapy, but are found in only about 50% of patients, mainly those with APL and Philadelphia chromosome-positive leukemias.
With driver mutations identified in the majority of patients, we can be hopeful that NGS negativity may be a useful clinical endpoint. In work presented at ASH 2016 by Bartlomiej M Getta, MBBS, of Memorial Sloan Kettering Cancer Center, New York, and his colleagues, patients with concordant MRD positivity by flow cytometry and NGS had inferior outcomes, even after allogeneic transplant, compared to patients with MRD positivity on one assay but not both.5 Nonetheless, caution should be taken in early adoption of NGS as a independent marker of MRD status for two main reasons: 1) False positives and lack of standardization make current interpretation difficult. 2) The presence of “preleukemic” clones remains enigmatic – and no matter the nomenclature used, can a DNMT3A or IDH-mutant clone really be deemed “clonal hematopoiesis of indeterminate potential” when a patient has already had clonal transformation?
Conversely, not all patients reported in the work by Klco2 and Getta ultimately relapse. Thus, while it would be preferred to clear all mutant clones, as a therapeutic goal this likely would subject many patients to unnecessary toxicity. One half of the patients reported by Getta were disease free at a year with concordant flow and NGS positive MRD while patients with NGS positivity alone had outcomes equivalent to those of MRD-negative patients, highlighting that certain persistent clones in NGS-only, MRD-positive patients might be amenable to immunotherapy, either with checkpoint inhibitors or allogeneic transplant. Insight into which clones remain quiescent and which are more sinister will require more investigation, but there does seem to be an additive role to NGS-positivity, whereby all MRD is not created equal and the precision and clinical utility of MRD status will likely take on nuanced nomenclature.
References
1. Jan, M. et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine 4, 149ra118, doi: 10.1126/scitranslmed.3004315 (2012).
2. Klco, J. M. et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015;314:811-22. doi: 10.1001/jama.2015.9643.
3. Wong, T. N. et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016;127:893-7. doi: 10.1182/blood-2015-10-677021 (2016).
4. Lane, A. A. et al. Results from Ongoing Phase II Trial of SL-401 As Consolidation Therapy in Patients with Acute Myeloid Leukemia (AML) in Remission with High Relapse Risk Including Minimal Residual Disease (MRD), Abstract 215, ASH 2016.
5. Getta, B. M. et al. Multicolor Flow Cytometry and Multi-Gene Next Generation Sequencing Are Complimentary and Highly Predictive for Relapse in Acute Myeloid Leukemia Following Allogeneic Hematopoietic Stem Cell Transplant, Abstract 834, ASH 2016.
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, New York, where he is a clinical instructor, on the staff of the leukemia service, and a clinical researcher in The Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
In hematologic malignancies, there is a deep and direct connection between each individual patient, that patient’s symptoms, the visible cells that cause the disease, and the direct measurements and assessments of those cells. The totality of these factors helps to determine the diagnosis and treatment plan. As a butterfly needle often is sufficient for obtaining a diagnostic tumor biopsy, it is not surprising that these same diagnostic techniques are now standardly being used to monitor disease response.
The techniques differ in their limits of detection, however. With sequencing depths able to reliably detect variant allele frequencies of less than 10%, even when patients’ overt leukemia may no longer be detectable, clinicians may be left to ponder what to do with persistent “preleukemic” or “rising clones.”1-3
These patients, now more appropriately stratified for risk of recurrence, are in desperate need of better care algorithms. Standard MRD assessment by flow cytometric analysis is able to detect less than 1 x 10-4 cells. While it can be applied to most patients, its sensitivity will likely be surpassed by new and emerging genomic assays. Real time quantitative polymerase chain reaction (RT-qPCR) and next generation sequencing (NGS) require a leukemia-specific abnormality but have the potential for far greater sensitivity with deeper sequencing techniques.
Long-term follow up data in acute promyelocytic leukemia (APL) provides the illustrative example where morphologic remission is not durable in the setting of a persistent PML-RARa transcript and therapeutic goals for PCR negativity irrespective of morphology are standard. Pathologic fusion proteins are ideal for marker-driven therapy, but are found in only about 50% of patients, mainly those with APL and Philadelphia chromosome-positive leukemias.
With driver mutations identified in the majority of patients, we can be hopeful that NGS negativity may be a useful clinical endpoint. In work presented at ASH 2016 by Bartlomiej M Getta, MBBS, of Memorial Sloan Kettering Cancer Center, New York, and his colleagues, patients with concordant MRD positivity by flow cytometry and NGS had inferior outcomes, even after allogeneic transplant, compared to patients with MRD positivity on one assay but not both.5 Nonetheless, caution should be taken in early adoption of NGS as a independent marker of MRD status for two main reasons: 1) False positives and lack of standardization make current interpretation difficult. 2) The presence of “preleukemic” clones remains enigmatic – and no matter the nomenclature used, can a DNMT3A or IDH-mutant clone really be deemed “clonal hematopoiesis of indeterminate potential” when a patient has already had clonal transformation?
Conversely, not all patients reported in the work by Klco2 and Getta ultimately relapse. Thus, while it would be preferred to clear all mutant clones, as a therapeutic goal this likely would subject many patients to unnecessary toxicity. One half of the patients reported by Getta were disease free at a year with concordant flow and NGS positive MRD while patients with NGS positivity alone had outcomes equivalent to those of MRD-negative patients, highlighting that certain persistent clones in NGS-only, MRD-positive patients might be amenable to immunotherapy, either with checkpoint inhibitors or allogeneic transplant. Insight into which clones remain quiescent and which are more sinister will require more investigation, but there does seem to be an additive role to NGS-positivity, whereby all MRD is not created equal and the precision and clinical utility of MRD status will likely take on nuanced nomenclature.
References
1. Jan, M. et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine 4, 149ra118, doi: 10.1126/scitranslmed.3004315 (2012).
2. Klco, J. M. et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015;314:811-22. doi: 10.1001/jama.2015.9643.
3. Wong, T. N. et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016;127:893-7. doi: 10.1182/blood-2015-10-677021 (2016).
4. Lane, A. A. et al. Results from Ongoing Phase II Trial of SL-401 As Consolidation Therapy in Patients with Acute Myeloid Leukemia (AML) in Remission with High Relapse Risk Including Minimal Residual Disease (MRD), Abstract 215, ASH 2016.
5. Getta, B. M. et al. Multicolor Flow Cytometry and Multi-Gene Next Generation Sequencing Are Complimentary and Highly Predictive for Relapse in Acute Myeloid Leukemia Following Allogeneic Hematopoietic Stem Cell Transplant, Abstract 834, ASH 2016.
Dr. Viny is with the Memorial Sloan-Kettering Cancer Center, New York, where he is a clinical instructor, on the staff of the leukemia service, and a clinical researcher in The Ross Levine Lab. Contact Dr. Viny at vinya@mskcc.org.
10 recommendations for the Cancer Moonshot
Responding to the Cancer Moonshot initiative, a panel of scientists, clinicians, patient advocates, and industry representatives has issued 10 recommendations for accelerating cancer research in an article published in Science.
The recommendations address:
• Development of a patient engagement network.
• Precise cataloging of tumor molecular changes.
• Analysis of samples already available from patients who have received the standard of care.

• Improvements for data sharing, access, and analysis.
• Development of models to understand how childhood cancers develop.
• Research to describe how fusion oncoproteins drive cancer development.
• Creation of a cancer immunotherapy clinical trials network.
• Systematic efforts to gather information on patient-reported outcomes.
• Implementation of evidence-based approaches to prevention.
The panel’s recommendations were presented to the National Cancer Advisory Board, the adviser to the National Cancer Institute. The ability to conduct research stemming from the panel’s recommendations will depend on whether, and how much, funding is approved by Congress.
Read the article here: http://science.sciencemag.org/content/early/2016/09/07/science.aai7862.full.
Responding to the Cancer Moonshot initiative, a panel of scientists, clinicians, patient advocates, and industry representatives has issued 10 recommendations for accelerating cancer research in an article published in Science.
The recommendations address:
• Development of a patient engagement network.
• Precise cataloging of tumor molecular changes.
• Analysis of samples already available from patients who have received the standard of care.
• Improvements for data sharing, access, and analysis.
• Development of models to understand how childhood cancers develop.
• Research to describe how fusion oncoproteins drive cancer development.
• Creation of a cancer immunotherapy clinical trials network.
• Systematic efforts to gather information on patient-reported outcomes.
• Implementation of evidence-based approaches to prevention.
The panel’s recommendations were presented to the National Cancer Advisory Board, the adviser to the National Cancer Institute. The ability to conduct research stemming from the panel’s recommendations will depend on whether, and how much, funding is approved by Congress.
Read the article here: http://science.sciencemag.org/content/early/2016/09/07/science.aai7862.full.
Responding to the Cancer Moonshot initiative, a panel of scientists, clinicians, patient advocates, and industry representatives has issued 10 recommendations for accelerating cancer research in an article published in Science.
The recommendations address:
• Development of a patient engagement network.
• Precise cataloging of tumor molecular changes.
• Analysis of samples already available from patients who have received the standard of care.
• Improvements for data sharing, access, and analysis.
• Development of models to understand how childhood cancers develop.
• Research to describe how fusion oncoproteins drive cancer development.
• Creation of a cancer immunotherapy clinical trials network.
• Systematic efforts to gather information on patient-reported outcomes.
• Implementation of evidence-based approaches to prevention.
The panel’s recommendations were presented to the National Cancer Advisory Board, the adviser to the National Cancer Institute. The ability to conduct research stemming from the panel’s recommendations will depend on whether, and how much, funding is approved by Congress.
Read the article here: http://science.sciencemag.org/content/early/2016/09/07/science.aai7862.full.

Dasatinib plus venetoclax shows promise in mouse model of Ph+ALL
The combination of dasatinib and venetoclax had a synergistic effect that was associated with lower toxicity than single-agent therapy, based on responses of primary Philadelphia chromosome–positive acute lymphoblastic leukemia (PH+ALL) samples in xenografted immunodeficient mice, according to Jessica T. Leonard, MD.
Dr. Leonard and her colleagues at Oregon Health and Science University in Portland demonstrated that the combination of venetoclax – a selective inhibitor of B cell lymphoma 2 – was highly synergistic with tyrosine kinase inhibitors in vitro. In the preclinical model of PH+ALL, a stepwise reduction in median inhibitory concentration of dasatinib was observed with increasing doses of venetoclax, as was decreased cell viability and induced apoptosis. Dasatinib – a breakpoint cluster region–Abelson kinase inhibitor – has an additional advantage of potentially overcoming venetoclax resistance by blocking a common mechanism of resistance to the agent, the investigators reported Aug. 31 in Science Translational Medicine (2016; 8[354]:354ra114).
The combination boosted antitumor activity against Ph+ALL cells grown in culture. In the mouse model of Ph+ALL, all of the mice in the combination dosing group remained alive during the 4-week treatment period. The combination therapy was well tolerated, and superior to either agent alone with respect to antileukemic efficacy.
The investigators focused on combining venetoclax with dasatinib because it is “the current backbone for the treatment of adult Ph+ALL ... These results lay the foundation for the testing of this combination in patients with Ph+ALL with the goal of improving treatment,” they concluded.
This study was supported in part by the Leukemia & Lymphoma Society and the Newman’s Own Foundation. Dr. Leonard reported having no other disclosures, but various coauthors reported receiving research support from, and/or serving as a consultant or scientific advisory board member to Genentech, the maker of venetoclax, and Bristol-Myers Squibb, the maker of dasatinib, as well as numerous other drug companies.
The combination of dasatinib and venetoclax had a synergistic effect that was associated with lower toxicity than single-agent therapy, based on responses of primary Philadelphia chromosome–positive acute lymphoblastic leukemia (PH+ALL) samples in xenografted immunodeficient mice, according to Jessica T. Leonard, MD.
Dr. Leonard and her colleagues at Oregon Health and Science University in Portland demonstrated that the combination of venetoclax – a selective inhibitor of B cell lymphoma 2 – was highly synergistic with tyrosine kinase inhibitors in vitro. In the preclinical model of PH+ALL, a stepwise reduction in median inhibitory concentration of dasatinib was observed with increasing doses of venetoclax, as was decreased cell viability and induced apoptosis. Dasatinib – a breakpoint cluster region–Abelson kinase inhibitor – has an additional advantage of potentially overcoming venetoclax resistance by blocking a common mechanism of resistance to the agent, the investigators reported Aug. 31 in Science Translational Medicine (2016; 8[354]:354ra114).
The combination boosted antitumor activity against Ph+ALL cells grown in culture. In the mouse model of Ph+ALL, all of the mice in the combination dosing group remained alive during the 4-week treatment period. The combination therapy was well tolerated, and superior to either agent alone with respect to antileukemic efficacy.
The investigators focused on combining venetoclax with dasatinib because it is “the current backbone for the treatment of adult Ph+ALL ... These results lay the foundation for the testing of this combination in patients with Ph+ALL with the goal of improving treatment,” they concluded.
This study was supported in part by the Leukemia & Lymphoma Society and the Newman’s Own Foundation. Dr. Leonard reported having no other disclosures, but various coauthors reported receiving research support from, and/or serving as a consultant or scientific advisory board member to Genentech, the maker of venetoclax, and Bristol-Myers Squibb, the maker of dasatinib, as well as numerous other drug companies.
The combination of dasatinib and venetoclax had a synergistic effect that was associated with lower toxicity than single-agent therapy, based on responses of primary Philadelphia chromosome–positive acute lymphoblastic leukemia (PH+ALL) samples in xenografted immunodeficient mice, according to Jessica T. Leonard, MD.
Dr. Leonard and her colleagues at Oregon Health and Science University in Portland demonstrated that the combination of venetoclax – a selective inhibitor of B cell lymphoma 2 – was highly synergistic with tyrosine kinase inhibitors in vitro. In the preclinical model of PH+ALL, a stepwise reduction in median inhibitory concentration of dasatinib was observed with increasing doses of venetoclax, as was decreased cell viability and induced apoptosis. Dasatinib – a breakpoint cluster region–Abelson kinase inhibitor – has an additional advantage of potentially overcoming venetoclax resistance by blocking a common mechanism of resistance to the agent, the investigators reported Aug. 31 in Science Translational Medicine (2016; 8[354]:354ra114).
The combination boosted antitumor activity against Ph+ALL cells grown in culture. In the mouse model of Ph+ALL, all of the mice in the combination dosing group remained alive during the 4-week treatment period. The combination therapy was well tolerated, and superior to either agent alone with respect to antileukemic efficacy.
The investigators focused on combining venetoclax with dasatinib because it is “the current backbone for the treatment of adult Ph+ALL ... These results lay the foundation for the testing of this combination in patients with Ph+ALL with the goal of improving treatment,” they concluded.
This study was supported in part by the Leukemia & Lymphoma Society and the Newman’s Own Foundation. Dr. Leonard reported having no other disclosures, but various coauthors reported receiving research support from, and/or serving as a consultant or scientific advisory board member to Genentech, the maker of venetoclax, and Bristol-Myers Squibb, the maker of dasatinib, as well as numerous other drug companies.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: The combination of dasatinib and venetoclax shows promise for the treatment of primary Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ALL) samples in xenografted immunodeficient mice and should be further evaluated for patient care.
Major finding: A stepwise reduction in median inhibitory concentration of dasatinib was seen with increasing doses of venetoclax.
Data source: In vitro and in vivo evaluation of BCL-2 inhibition in combination with kinase inhibition in a murine model of Ph+ALL.
Disclosures: This study was supported in part by the Leukemia & Lymphoma Society and the Newman’s Own Foundation. Dr. Leonard reported having no other disclosures, but various coauthors reported receiving research support from, and/or serving as a consultant or scientific advisory board member to Genentech, the maker of venetoclax, and Bristol-Myers Squibb, the maker of dasatinib, as well as numerous other drug companies.
The liver drives RBC elimination, iron recycling
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
FROM NATURE MEDICINE
Dematin key to erythrocyte membrane stability in mice
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Dematin is newly recognized as a protein crucial to the integrity of red blood cell membranes.
Major finding: Dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia.
Data source: Studies in a newly created mouse model designed to lack dematin.
Disclosures: The researchers had no relevant financial disclosures.
CUDC-907 enters phase II for relapsed or refractory lymphoma and multiple myeloma
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
FROM THE LANCET ONCOLOGY
Key clinical point: The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule.
Major finding: Two complete responses and three partial responses were seen in the subgroup of nine patients with diffuse large B-cell lymphoma; three occurred in the five patients with transformed follicular disease.
Data source: A dose-escalation study involving 44 patients at four cancer centers.
Disclosures: The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
Nanoparticles deliver Aurora kinase inhibitor with increased safety and efficacy
Using nanoparticles to encapsulate an Aurora B kinase inhibitor improved the efficacy and tolerability of the drug and allowed less frequent dosing in preclinical models, according to researchers.
“The AZD2811 nanoparticles identified in this study have the potential to increase efficacy at tolerable doses using a more convenient dosing regimen, which may in turn extend the utility of Aurora B kinase inhibition to a broader range of hematological and solid tumor cancer indications,” wrote Susan Ashton of AstraZeneca, and her colleagues (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad2355).
“The improved bone marrow profile observed with slow-releasing nanoparticles may enable efficacious combination treatments” with chemotherapy, radiotherapy, or poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors.
The study was undertaken because a free-drug version of the agent, known as AZD1152, had led to a significant improvement in the complete response rate of acute myeloid leukemia compared to standard of care in a phase II trial. Efficacy, however, was associated with major toxicities, including myelosuppression. Further, AZD1152 had to be administered as a 7-day continuous intravenous infusion.
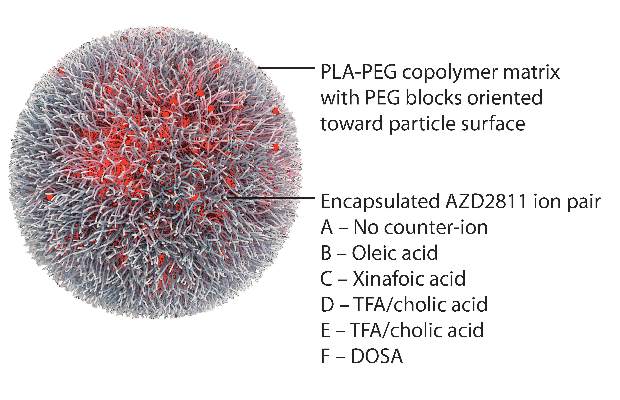
By using the Accurin nanoparticle platform to vary drug release kinetics, the researchers devised a formulation to maximize the therapeutic effect of the kinase inhibitor while sparing healthy tissue. AZD1152 is a water-soluble prodrug of AZD2811, which the researchers used to develop their the nanoparticle formulation.
AZD2811 was encapsulated in polymeric nanoparticles termed Accurins, which are composed of block copolymers of poly-D,L-lactide (PLA) and poly(ethylene glycol) (PEG). Accurins accumulate in tumors, increasing the drug’s concentration and duration of exposure to the cancer cells. Organic acid counterions were used to increase encapsulation efficiency and decrease the release rate of AZD2811.
“We identified a formulation profile that could deliver active drug for more than 1 week, resulting in prolonged target inhibition in tumor tissue together with improved preclinical efficacy and therapeutic index over the AZD1152 prodrug in several animal models,” they wrote.
In nude rats bearing human colorectal adenocarcinoma SW620 xenografts, the nanoparticles inhibited kinase over a 96-hour time course, while the free drug resulted in complete enzyme recovery at 24 hours. Nanoparticles inhibited tumor growth by over 90%, compared with 58% for the free drug at twice the dose, and showed little toxicity as evidenced by stable body weight. Nanoparticles were retained in the tumor xenografts for up to 6 days, while the free drug was undetected in tumors 24 hours after administration.
“Although we selected a lead formulation using a tumor model (SW620) that supported the AZD1152 program – and, as such, we had extensive comparator data from which to benchmark the tolerability, PD, and efficacy of candidate nanoparticles – the model is subject to the known limitations of xenografted human tumor cell lines in assessing therapeutic candidates in oncology. Moreover, although rat bone marrow is commonly used to model myelotoxicity in humans, interrogation of the nanoparticle dose and schedule in patients may be required to achieve optimal clinical results,” they concluded.
AstraZeneca funded the study. Dr. Ashton and several coauthors are current or former employees and shareholders of AstraZeneca or BIND. The companies are developing the drug and technologies.
By encapsulating an Aurora B kinase inhibitor in Accurin particles, the researchers appear to have succeeded in enhancing the drug’s therapeutic activity and safety in mouse xenograft models. It remains to be seen how widely applicable this technology will be, and whether these results will be replicated in patients as the AZD1151-hQPA Accurin formulation heads toward first-in-human clinical trials. If the Accurin technology can solve the pharmacokinetic and toxicity issues for Aurora kinase inhibitors, it will likely also be applicable to other compounds that have encountered similar difficulties in clinical development and will add a formulation approach to address very meaningful and challenging issues that face many new molecules during clinical development.
David J. Bearss, Ph.D., is the chief executive officer of Tolero Pharmaceutical, Lehi, Utah. His remarks were part of an editorial accompanying the report in Science Translational Medicine (2016 Feb 10. doi: 10.1126/scitranslmed.aaf1417).
By encapsulating an Aurora B kinase inhibitor in Accurin particles, the researchers appear to have succeeded in enhancing the drug’s therapeutic activity and safety in mouse xenograft models. It remains to be seen how widely applicable this technology will be, and whether these results will be replicated in patients as the AZD1151-hQPA Accurin formulation heads toward first-in-human clinical trials. If the Accurin technology can solve the pharmacokinetic and toxicity issues for Aurora kinase inhibitors, it will likely also be applicable to other compounds that have encountered similar difficulties in clinical development and will add a formulation approach to address very meaningful and challenging issues that face many new molecules during clinical development.
David J. Bearss, Ph.D., is the chief executive officer of Tolero Pharmaceutical, Lehi, Utah. His remarks were part of an editorial accompanying the report in Science Translational Medicine (2016 Feb 10. doi: 10.1126/scitranslmed.aaf1417).
By encapsulating an Aurora B kinase inhibitor in Accurin particles, the researchers appear to have succeeded in enhancing the drug’s therapeutic activity and safety in mouse xenograft models. It remains to be seen how widely applicable this technology will be, and whether these results will be replicated in patients as the AZD1151-hQPA Accurin formulation heads toward first-in-human clinical trials. If the Accurin technology can solve the pharmacokinetic and toxicity issues for Aurora kinase inhibitors, it will likely also be applicable to other compounds that have encountered similar difficulties in clinical development and will add a formulation approach to address very meaningful and challenging issues that face many new molecules during clinical development.
David J. Bearss, Ph.D., is the chief executive officer of Tolero Pharmaceutical, Lehi, Utah. His remarks were part of an editorial accompanying the report in Science Translational Medicine (2016 Feb 10. doi: 10.1126/scitranslmed.aaf1417).
Using nanoparticles to encapsulate an Aurora B kinase inhibitor improved the efficacy and tolerability of the drug and allowed less frequent dosing in preclinical models, according to researchers.
“The AZD2811 nanoparticles identified in this study have the potential to increase efficacy at tolerable doses using a more convenient dosing regimen, which may in turn extend the utility of Aurora B kinase inhibition to a broader range of hematological and solid tumor cancer indications,” wrote Susan Ashton of AstraZeneca, and her colleagues (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad2355).
“The improved bone marrow profile observed with slow-releasing nanoparticles may enable efficacious combination treatments” with chemotherapy, radiotherapy, or poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors.
The study was undertaken because a free-drug version of the agent, known as AZD1152, had led to a significant improvement in the complete response rate of acute myeloid leukemia compared to standard of care in a phase II trial. Efficacy, however, was associated with major toxicities, including myelosuppression. Further, AZD1152 had to be administered as a 7-day continuous intravenous infusion.
By using the Accurin nanoparticle platform to vary drug release kinetics, the researchers devised a formulation to maximize the therapeutic effect of the kinase inhibitor while sparing healthy tissue. AZD1152 is a water-soluble prodrug of AZD2811, which the researchers used to develop their the nanoparticle formulation.
AZD2811 was encapsulated in polymeric nanoparticles termed Accurins, which are composed of block copolymers of poly-D,L-lactide (PLA) and poly(ethylene glycol) (PEG). Accurins accumulate in tumors, increasing the drug’s concentration and duration of exposure to the cancer cells. Organic acid counterions were used to increase encapsulation efficiency and decrease the release rate of AZD2811.
“We identified a formulation profile that could deliver active drug for more than 1 week, resulting in prolonged target inhibition in tumor tissue together with improved preclinical efficacy and therapeutic index over the AZD1152 prodrug in several animal models,” they wrote.
In nude rats bearing human colorectal adenocarcinoma SW620 xenografts, the nanoparticles inhibited kinase over a 96-hour time course, while the free drug resulted in complete enzyme recovery at 24 hours. Nanoparticles inhibited tumor growth by over 90%, compared with 58% for the free drug at twice the dose, and showed little toxicity as evidenced by stable body weight. Nanoparticles were retained in the tumor xenografts for up to 6 days, while the free drug was undetected in tumors 24 hours after administration.
“Although we selected a lead formulation using a tumor model (SW620) that supported the AZD1152 program – and, as such, we had extensive comparator data from which to benchmark the tolerability, PD, and efficacy of candidate nanoparticles – the model is subject to the known limitations of xenografted human tumor cell lines in assessing therapeutic candidates in oncology. Moreover, although rat bone marrow is commonly used to model myelotoxicity in humans, interrogation of the nanoparticle dose and schedule in patients may be required to achieve optimal clinical results,” they concluded.
AstraZeneca funded the study. Dr. Ashton and several coauthors are current or former employees and shareholders of AstraZeneca or BIND. The companies are developing the drug and technologies.
Using nanoparticles to encapsulate an Aurora B kinase inhibitor improved the efficacy and tolerability of the drug and allowed less frequent dosing in preclinical models, according to researchers.
“The AZD2811 nanoparticles identified in this study have the potential to increase efficacy at tolerable doses using a more convenient dosing regimen, which may in turn extend the utility of Aurora B kinase inhibition to a broader range of hematological and solid tumor cancer indications,” wrote Susan Ashton of AstraZeneca, and her colleagues (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad2355).
“The improved bone marrow profile observed with slow-releasing nanoparticles may enable efficacious combination treatments” with chemotherapy, radiotherapy, or poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors.
The study was undertaken because a free-drug version of the agent, known as AZD1152, had led to a significant improvement in the complete response rate of acute myeloid leukemia compared to standard of care in a phase II trial. Efficacy, however, was associated with major toxicities, including myelosuppression. Further, AZD1152 had to be administered as a 7-day continuous intravenous infusion.
By using the Accurin nanoparticle platform to vary drug release kinetics, the researchers devised a formulation to maximize the therapeutic effect of the kinase inhibitor while sparing healthy tissue. AZD1152 is a water-soluble prodrug of AZD2811, which the researchers used to develop their the nanoparticle formulation.
AZD2811 was encapsulated in polymeric nanoparticles termed Accurins, which are composed of block copolymers of poly-D,L-lactide (PLA) and poly(ethylene glycol) (PEG). Accurins accumulate in tumors, increasing the drug’s concentration and duration of exposure to the cancer cells. Organic acid counterions were used to increase encapsulation efficiency and decrease the release rate of AZD2811.
“We identified a formulation profile that could deliver active drug for more than 1 week, resulting in prolonged target inhibition in tumor tissue together with improved preclinical efficacy and therapeutic index over the AZD1152 prodrug in several animal models,” they wrote.
In nude rats bearing human colorectal adenocarcinoma SW620 xenografts, the nanoparticles inhibited kinase over a 96-hour time course, while the free drug resulted in complete enzyme recovery at 24 hours. Nanoparticles inhibited tumor growth by over 90%, compared with 58% for the free drug at twice the dose, and showed little toxicity as evidenced by stable body weight. Nanoparticles were retained in the tumor xenografts for up to 6 days, while the free drug was undetected in tumors 24 hours after administration.
“Although we selected a lead formulation using a tumor model (SW620) that supported the AZD1152 program – and, as such, we had extensive comparator data from which to benchmark the tolerability, PD, and efficacy of candidate nanoparticles – the model is subject to the known limitations of xenografted human tumor cell lines in assessing therapeutic candidates in oncology. Moreover, although rat bone marrow is commonly used to model myelotoxicity in humans, interrogation of the nanoparticle dose and schedule in patients may be required to achieve optimal clinical results,” they concluded.
AstraZeneca funded the study. Dr. Ashton and several coauthors are current or former employees and shareholders of AstraZeneca or BIND. The companies are developing the drug and technologies.
Key clinical point: Aurora B kinase inhibitor nanoparticles displayed accumulation and retention in tumors with improved efficacy and minimal bone marrow pathology in animal models.
Major finding: Nanoparticles inhibited tumor growth by over 90%, compared with 58% for the free drug at twice the dose, and showed little toxicity; the free drug was undetected in tumors 24 hours after administration, and nanoparticle-delivered drug was detectable up to 6 days.
Data sources: Nude rats and nude mice bearing human colorectal adenocarcinoma SW620 xenografts.
Disclosures: AstraZeneca funded the study. Dr. Ashton and several coauthors are current or former employees and shareholders of AstraZeneca or BIND. The companies are developing the drug and technologies.
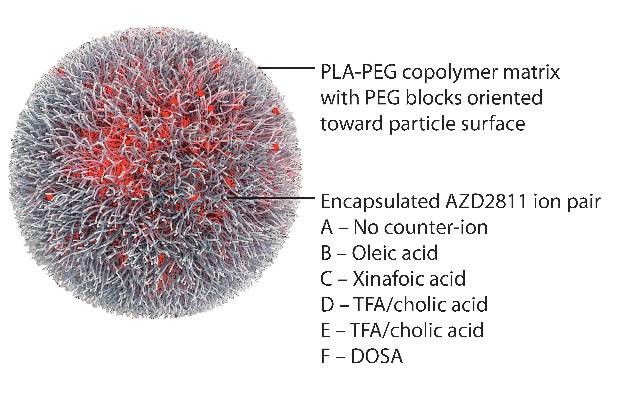
Novel test detects low levels of residual CML
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.

Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
FROM THE JOURNAL OF MOLECULAR DIAGNOSTICS
Key clinical point: A novel test may predict which CML patients in remission can safely stop TKI therapy.
Major finding: Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%).
Data source: DNA-based dPCR measures in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Disclosures: The researchers had no relevant disclosures.
