User login
What is the best approach to a high systolic pulmonary artery pressure on echocardiography?
The incidental finding of high systolic pulmonary artery pressure on echocardiography is common. What we should do about it varies according to clinical presentation, comorbidities, and results of other tests, including assessment of the right ventricle. Thus, the optimal approach ranges from no further investigation to right heart catheterization and, in some cases, referral to a pulmonary hypertension center.
THE TWO MEASUREMENTS COMPARED
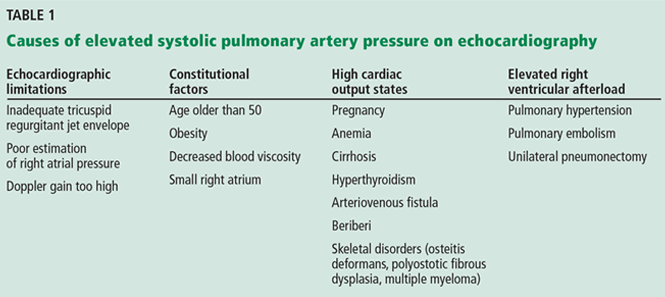
Although it raises concern, the finding of high systolic pulmonary artery pressure is not enough to diagnose pulmonary hypertension. In fact, several other conditions are associated with high systolic pulmonary artery pressure on echocardiography (Table 1). The diagnosis must be confirmed with right heart catheterization.1
Echocardiography provides an estimate of the systolic pulmonary artery pressure that is calculated from other values, whereas right heart catheterization gives a direct measurement of the mean pulmonary artery pressure, which is necessary for diagnosing pulmonary hypertension. The two values are correlated, but the differences are noteworthy.
WHAT IS PULMONARY HYPERTENSION?
Pulmonary hypertension is defined by a resting mean pulmonary artery pressure 25 mm Hg or greater during right heart catheterization.1 The large number of conditions associated with pulmonary hypertension can be divided into five groups2:
- Group 1, pulmonary artery hypertension
- Group 2, pulmonary hypertension associated with left heart disease
- Group 3, pulmonary hypertension due to chronic lung disease or hypoxia
- Group 4, chronic thromboembolic pulmonary hypertension
- Group 5, pulmonary hypertension due to unclear multifactorial mechanisms.2
Pulmonary artery hypertension (group 1) is a syndrome characterized by a restricted flow of small pulmonary arteries that can be idiopathic, heritable, or induced by anorexigens, connective tissue disease, congenital heart disease, portal hypertension, human immunodeficiency virus (HIV), or schistosomiasis.2,3 In spite of significant advances in therapy in the last 3 decades, pulmonary artery hypertension continues to lead to right heart failure and death,4 and the diagnosis has adverse prognostic implications. Therefore, it is essential to be attentive when reviewing the echocardiogram, since an elevated systolic pulmonary artery pressure may be an important clue to pulmonary hypertension.
ESTIMATED PRESSURE: HOW HIGH IS TOO HIGH?
There is no consensus on the optimal cutoff of echocardiographic systolic pulmonary artery pressure to trigger a further evaluation for pulmonary hypertension.
A retrospective evaluation of nearly 16,000 normal echocardiograms found that the 95% upper limit for systolic pulmonary artery pressure was 37 mm Hg.5
European guidelines6 propose that pulmonary hypertension is unlikely if the estimated systolic pulmonary artery pressure is 36 mm Hg or lower, possible if it is 37 to 50 mm Hg, and likely if it is higher than 50 mm Hg.6
The 2009 consensus document of the American College of Cardiology Foundation and American Heart Association3 recommends a systolic pulmonary artery pressure greater than 40 mm Hg as the threshold to suggest further evaluation in a patient with unexplained dyspnea.
Converting the systolic pulmonary artery pressure to the mean pressure
Although not validated to use with echocardiography, the most accurate estimate of mean pulmonary artery pressure was shown in one study7 to be obtained with the equation:
0.61 × systolic pulmonary artery pressure
+ 2 mm Hg
Using this formula, a systolic pulmonary artery pressure of 37 mm Hg would correspond to a mean pulmonary artery pressure of 24.6 mm Hg. A systolic pulmonary artery pressure of 40 mm Hg would correspond to a mean pulmonary artery pressure of 26.4 mm Hg.
Estimated systolic pulmonary artery pressure depends on several variables
Systolic pulmonary artery pressure is estimated using the simplified Bernoulli equation8:
4 × tricuspid regurgitation jet velocity2 (m/s)
+ right atrial pressure (mm Hg)
Tricuspid regurgitation is present in over 75% of the normal population. The regurgitation velocity across the tricuspid valve must be measured to estimate the pressure gradient between the right ventricle and the right atrium. The right atrial pressure is estimated from the diameter of the inferior vena cava and the degree of inspiratory collapse with the sniff test. As the right atrial pressure increases, the inferior vena cava dilates and inspiratory collapse decreases.8 If there is no gradient across the right ventricular outflow tract or pulmonary valve, the right ventricular systolic pressure is equal to the systolic pulmonary artery pressure.
Since tricuspid regurgitation velocity is squared and then multiplied by 4, small deviations of this measurement lead to markedly different systolic pulmonary artery pressure values. To avoid this problem, the tricuspid regurgitation velocity needs to be looked at in multiple echocardiographic views to find the best alignment with the flow and an adequate envelope.
Many causes of high estimated systolic pulmonary artery pressure
Table 1 shows conditions associated with a high estimated systolic pulmonary artery pressure. Echocardiographic limitations, constitutional factors, and high cardiac output states can lead to an apparent elevation in systolic pulmonary artery pressure, which is not confirmed later during right heart catheterization.
Systolic pulmonary artery pressure increases with age and body mass index as a result of worsening left ventricular diastolic dysfunction.8 In fact, an estimated pressure greater than 40 mm Hg is found5 in 6% of people over age 50 and in 5% of people with a body mass index greater than 30 kg/m2. It can also be high in conditions in which there is an increase in cardiac output, such as pregnancy, anemia (sickle cell disease, thalassemia), cirrhosis, and arteriovenous fistula.
The estimated systolic value often differs from the measured value
Studies have compared the systolic pulmonary artery pressure measured during right heart catheterization with the estimated value on echocardiography.9,10 These studies noted a reasonable degree of agreement between the tests but a substantial variability.
Both underestimation and overestimation of the systolic pulmonary artery pressure by echocardiography were common, with 95% limits of agreement ranging from minus 40 mm Hg to plus 40 mm Hg.9,10 A difference of plus or minus 10 mm Hg in systolic pulmonary artery pressure between echocardiography and catheterization was observed in 48% to 51% of patients with pulmonary hypertension, particularly in those with higher systolic pulmonary artery pressure.9,10
An important reason for overestimation of systolic pulmonary artery pressure is the inaccurate estimation of the right atrial pressure by echocardiography.9,10 Indeed, this factor may account for half of the cases in which the systolic pulmonary artery pressure is overestimated.10 Although the traditional methods to estimate the right atrial pressure have been revisited,8,11 this estimation is less reliable for intermediate pressure values, for patients on mechanical ventilation, and for young athletes.8
Other explanations for the variability between measured and estimated systolic pulmonary artery pressure include suboptimal alignment between the Doppler beam and the regurgitant jet, severe tricuspid regurgitation, arrhythmias, and limitations inherent to the simplified Bernoulli equation.12 The estimated value is particularly inaccurate in patients with advanced lung disease, possibly owing to lung hyperinflation and alteration in the thoracic cavity and position of the heart—all factors that limit visualization and measurement of the tricuspid regurgitant jet.13
OTHER SIGNS OF PULMONARY HYPERTENSION ON ECHOCARDIOGRAPHY
Echocardiography provides information that is useful in assessing the accuracy of the estimated systolic pulmonary artery pressure, particularly right ventricular size and function.
As pulmonary hypertension progresses, the right ventricle dilates, and its function is compromised. Therefore, it is important to determine the right ventricular size and function by using objective echocardiographic findings such as right ventricular diameters (basal, mid, apical) and area, right ventricular fractional area change, tricuspid annular plane systolic excursion, myocardial performance index, and the pulsed tissue Doppler tricuspid annular peak systolic excursion velocity.8
Other echocardiographic features that suggest pulmonary hypertension include a dilated right atrial area, flattening of the interventricular septum, notching of the right ventricular outflow tract flow, and dilation of the main pulmonary artery. Interestingly, left ventricular diastolic dysfunction of the impaired relaxation type (grade I) is commonly observed in pulmonary hypertension14; however, more advanced degrees of diastolic dysfunction, ie, pseudonormalization (grade II) or restrictive left ventricular filling (grade III),15 particularly when associated with a left atrial enlargement, suggest pulmonary hypertension associated with left heart disease and not pulmonary artery hypertension.
WHAT TO DO IF ECHOCARDIOGRAPHY INDICATES PULMONARY HYPERTENSION
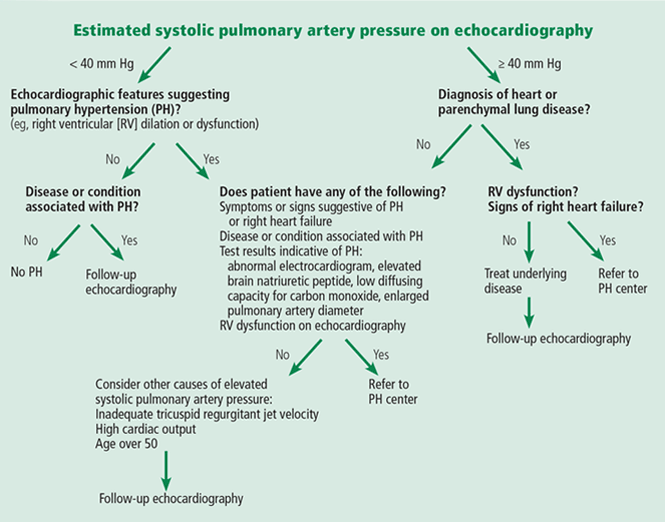
An algorithm showing the approach to an elevated systolic pulmonary artery pressure on echocardiography is presented in Figure 1.
In the appropriate clinical setting, if the systolic pulmonary artery pressure is 40 mm Hg or greater or if other echocardiographic variables suggest pulmonary hypertension, our practice is to proceed with right heart catheterization.
Clinical variables that suggest pulmonary hypertension include progressive dyspnea, chest pain, presyncope-syncope, lower extremity edema, hepatomegaly, jugular vein distention, hepatojugular reflux, sternal heave, loud second heart sound (P2), murmur of tricuspid or pulmonary regurgitation, and right ventricular third heart sound.16 These are of particular interest when associated with conditions known to cause pulmonary hypertension,2such as connective tissue disease, portal hypertension, congenital heart disease, HIV infection, and certain drugs and toxins.
Other tests that raise suspicion of pulmonary hypertension are an electrocardiogram suggesting a dilated right atrium or ventricle, an elevated brain natriuretic peptide level, a low carbon monoxide diffusing capacity on pulmonary function testing, and an enlarged pulmonary artery diameter on imaging.
Given the high prevalence of pulmonary hypertension, the Fifth World Symposium on Pulmonary Hypertension recommended first considering heart or parenchymal lung disease when an echocardiogram suggests pulmonary hypertension.6 If there are signs of severe pulmonary hypertension or right ventricular dysfunction, referral to a center specializing in pulmonary hypertension is recommended. Referral is also appropriate when there is no major heart or lung disease and the echocardiogram shows an elevated systolic pulmonary artery pressure, particularly when the clinical presentation or results of other testing suggest pulmonary hypertension.
TAKE-HOME POINTS
In the appropriate context, a high systolic pulmonary artery pressure on echocardiography suggests pulmonary hypertension, but right heart catheterization is needed to confirm the diagnosis. Estimating the systolic pulmonary artery pressure with echocardiography has limitations, including false-positive results, predominantly when the pretest probability of pulmonary hypertension is low.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D42–D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D34–D41.
- McLaughlin VV, Archer SL, Badesch DB, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009; 53:1573–1619.
- Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013; 188:365–369.
- McQuillan BM, Picard MH, Leavitt M, Weyman AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation 2001; 104:2797–2802.
- Galiè N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30:2493–2537.
- Chemla D, Castelain V, Provencher S, Humbert M, Simonneau G, Herve P. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest 2009; 135:760–768.
- Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 2010; 23:685–713.
- Rich JD, Shah SJ, Swamy RS, Kamp A, Rich S. Inaccuracy of Doppler echocardiographic estimates of pulmonary artery pressures in patients with pulmonary hypertension: implications for clinical practice. Chest 2011; 139:988–993.
- Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med 2009; 179:615–621.
- Brennan JM, Blair JE, Goonewardena S, et al. Reappraisal of the use of inferior vena cava for estimating right atrial pressure. J Am Soc Echocardiogr 2007; 20:857–861.
- Giardini A, Tacy TA. Non-invasive estimation of pressure gradients in regurgitant jets: an overdue consideration. Eur J Echocardiogr 2008; 9:578–584.
- Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003; 167:735–740.
- Tonelli AR, Plana JC, Heresi GA, Dweik RA. Prevalence and prognostic value of left ventricular diastolic dysfunction in idiopathic and heritable pulmonary arterial hypertension. Chest 2012; 141:1457–1465.
- Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009; 22:107–133.
- Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43(suppl S):40S–47S.
The incidental finding of high systolic pulmonary artery pressure on echocardiography is common. What we should do about it varies according to clinical presentation, comorbidities, and results of other tests, including assessment of the right ventricle. Thus, the optimal approach ranges from no further investigation to right heart catheterization and, in some cases, referral to a pulmonary hypertension center.
THE TWO MEASUREMENTS COMPARED
Although it raises concern, the finding of high systolic pulmonary artery pressure is not enough to diagnose pulmonary hypertension. In fact, several other conditions are associated with high systolic pulmonary artery pressure on echocardiography (Table 1). The diagnosis must be confirmed with right heart catheterization.1
Echocardiography provides an estimate of the systolic pulmonary artery pressure that is calculated from other values, whereas right heart catheterization gives a direct measurement of the mean pulmonary artery pressure, which is necessary for diagnosing pulmonary hypertension. The two values are correlated, but the differences are noteworthy.
WHAT IS PULMONARY HYPERTENSION?
Pulmonary hypertension is defined by a resting mean pulmonary artery pressure 25 mm Hg or greater during right heart catheterization.1 The large number of conditions associated with pulmonary hypertension can be divided into five groups2:
- Group 1, pulmonary artery hypertension
- Group 2, pulmonary hypertension associated with left heart disease
- Group 3, pulmonary hypertension due to chronic lung disease or hypoxia
- Group 4, chronic thromboembolic pulmonary hypertension
- Group 5, pulmonary hypertension due to unclear multifactorial mechanisms.2
Pulmonary artery hypertension (group 1) is a syndrome characterized by a restricted flow of small pulmonary arteries that can be idiopathic, heritable, or induced by anorexigens, connective tissue disease, congenital heart disease, portal hypertension, human immunodeficiency virus (HIV), or schistosomiasis.2,3 In spite of significant advances in therapy in the last 3 decades, pulmonary artery hypertension continues to lead to right heart failure and death,4 and the diagnosis has adverse prognostic implications. Therefore, it is essential to be attentive when reviewing the echocardiogram, since an elevated systolic pulmonary artery pressure may be an important clue to pulmonary hypertension.
ESTIMATED PRESSURE: HOW HIGH IS TOO HIGH?
There is no consensus on the optimal cutoff of echocardiographic systolic pulmonary artery pressure to trigger a further evaluation for pulmonary hypertension.
A retrospective evaluation of nearly 16,000 normal echocardiograms found that the 95% upper limit for systolic pulmonary artery pressure was 37 mm Hg.5
European guidelines6 propose that pulmonary hypertension is unlikely if the estimated systolic pulmonary artery pressure is 36 mm Hg or lower, possible if it is 37 to 50 mm Hg, and likely if it is higher than 50 mm Hg.6
The 2009 consensus document of the American College of Cardiology Foundation and American Heart Association3 recommends a systolic pulmonary artery pressure greater than 40 mm Hg as the threshold to suggest further evaluation in a patient with unexplained dyspnea.
Converting the systolic pulmonary artery pressure to the mean pressure
Although not validated to use with echocardiography, the most accurate estimate of mean pulmonary artery pressure was shown in one study7 to be obtained with the equation:
0.61 × systolic pulmonary artery pressure
+ 2 mm Hg
Using this formula, a systolic pulmonary artery pressure of 37 mm Hg would correspond to a mean pulmonary artery pressure of 24.6 mm Hg. A systolic pulmonary artery pressure of 40 mm Hg would correspond to a mean pulmonary artery pressure of 26.4 mm Hg.
Estimated systolic pulmonary artery pressure depends on several variables
Systolic pulmonary artery pressure is estimated using the simplified Bernoulli equation8:
4 × tricuspid regurgitation jet velocity2 (m/s)
+ right atrial pressure (mm Hg)
Tricuspid regurgitation is present in over 75% of the normal population. The regurgitation velocity across the tricuspid valve must be measured to estimate the pressure gradient between the right ventricle and the right atrium. The right atrial pressure is estimated from the diameter of the inferior vena cava and the degree of inspiratory collapse with the sniff test. As the right atrial pressure increases, the inferior vena cava dilates and inspiratory collapse decreases.8 If there is no gradient across the right ventricular outflow tract or pulmonary valve, the right ventricular systolic pressure is equal to the systolic pulmonary artery pressure.
Since tricuspid regurgitation velocity is squared and then multiplied by 4, small deviations of this measurement lead to markedly different systolic pulmonary artery pressure values. To avoid this problem, the tricuspid regurgitation velocity needs to be looked at in multiple echocardiographic views to find the best alignment with the flow and an adequate envelope.
Many causes of high estimated systolic pulmonary artery pressure
Table 1 shows conditions associated with a high estimated systolic pulmonary artery pressure. Echocardiographic limitations, constitutional factors, and high cardiac output states can lead to an apparent elevation in systolic pulmonary artery pressure, which is not confirmed later during right heart catheterization.
Systolic pulmonary artery pressure increases with age and body mass index as a result of worsening left ventricular diastolic dysfunction.8 In fact, an estimated pressure greater than 40 mm Hg is found5 in 6% of people over age 50 and in 5% of people with a body mass index greater than 30 kg/m2. It can also be high in conditions in which there is an increase in cardiac output, such as pregnancy, anemia (sickle cell disease, thalassemia), cirrhosis, and arteriovenous fistula.
The estimated systolic value often differs from the measured value
Studies have compared the systolic pulmonary artery pressure measured during right heart catheterization with the estimated value on echocardiography.9,10 These studies noted a reasonable degree of agreement between the tests but a substantial variability.
Both underestimation and overestimation of the systolic pulmonary artery pressure by echocardiography were common, with 95% limits of agreement ranging from minus 40 mm Hg to plus 40 mm Hg.9,10 A difference of plus or minus 10 mm Hg in systolic pulmonary artery pressure between echocardiography and catheterization was observed in 48% to 51% of patients with pulmonary hypertension, particularly in those with higher systolic pulmonary artery pressure.9,10
An important reason for overestimation of systolic pulmonary artery pressure is the inaccurate estimation of the right atrial pressure by echocardiography.9,10 Indeed, this factor may account for half of the cases in which the systolic pulmonary artery pressure is overestimated.10 Although the traditional methods to estimate the right atrial pressure have been revisited,8,11 this estimation is less reliable for intermediate pressure values, for patients on mechanical ventilation, and for young athletes.8
Other explanations for the variability between measured and estimated systolic pulmonary artery pressure include suboptimal alignment between the Doppler beam and the regurgitant jet, severe tricuspid regurgitation, arrhythmias, and limitations inherent to the simplified Bernoulli equation.12 The estimated value is particularly inaccurate in patients with advanced lung disease, possibly owing to lung hyperinflation and alteration in the thoracic cavity and position of the heart—all factors that limit visualization and measurement of the tricuspid regurgitant jet.13
OTHER SIGNS OF PULMONARY HYPERTENSION ON ECHOCARDIOGRAPHY
Echocardiography provides information that is useful in assessing the accuracy of the estimated systolic pulmonary artery pressure, particularly right ventricular size and function.
As pulmonary hypertension progresses, the right ventricle dilates, and its function is compromised. Therefore, it is important to determine the right ventricular size and function by using objective echocardiographic findings such as right ventricular diameters (basal, mid, apical) and area, right ventricular fractional area change, tricuspid annular plane systolic excursion, myocardial performance index, and the pulsed tissue Doppler tricuspid annular peak systolic excursion velocity.8
Other echocardiographic features that suggest pulmonary hypertension include a dilated right atrial area, flattening of the interventricular septum, notching of the right ventricular outflow tract flow, and dilation of the main pulmonary artery. Interestingly, left ventricular diastolic dysfunction of the impaired relaxation type (grade I) is commonly observed in pulmonary hypertension14; however, more advanced degrees of diastolic dysfunction, ie, pseudonormalization (grade II) or restrictive left ventricular filling (grade III),15 particularly when associated with a left atrial enlargement, suggest pulmonary hypertension associated with left heart disease and not pulmonary artery hypertension.
WHAT TO DO IF ECHOCARDIOGRAPHY INDICATES PULMONARY HYPERTENSION
An algorithm showing the approach to an elevated systolic pulmonary artery pressure on echocardiography is presented in Figure 1.
In the appropriate clinical setting, if the systolic pulmonary artery pressure is 40 mm Hg or greater or if other echocardiographic variables suggest pulmonary hypertension, our practice is to proceed with right heart catheterization.
Clinical variables that suggest pulmonary hypertension include progressive dyspnea, chest pain, presyncope-syncope, lower extremity edema, hepatomegaly, jugular vein distention, hepatojugular reflux, sternal heave, loud second heart sound (P2), murmur of tricuspid or pulmonary regurgitation, and right ventricular third heart sound.16 These are of particular interest when associated with conditions known to cause pulmonary hypertension,2such as connective tissue disease, portal hypertension, congenital heart disease, HIV infection, and certain drugs and toxins.
Other tests that raise suspicion of pulmonary hypertension are an electrocardiogram suggesting a dilated right atrium or ventricle, an elevated brain natriuretic peptide level, a low carbon monoxide diffusing capacity on pulmonary function testing, and an enlarged pulmonary artery diameter on imaging.
Given the high prevalence of pulmonary hypertension, the Fifth World Symposium on Pulmonary Hypertension recommended first considering heart or parenchymal lung disease when an echocardiogram suggests pulmonary hypertension.6 If there are signs of severe pulmonary hypertension or right ventricular dysfunction, referral to a center specializing in pulmonary hypertension is recommended. Referral is also appropriate when there is no major heart or lung disease and the echocardiogram shows an elevated systolic pulmonary artery pressure, particularly when the clinical presentation or results of other testing suggest pulmonary hypertension.
TAKE-HOME POINTS
In the appropriate context, a high systolic pulmonary artery pressure on echocardiography suggests pulmonary hypertension, but right heart catheterization is needed to confirm the diagnosis. Estimating the systolic pulmonary artery pressure with echocardiography has limitations, including false-positive results, predominantly when the pretest probability of pulmonary hypertension is low.
The incidental finding of high systolic pulmonary artery pressure on echocardiography is common. What we should do about it varies according to clinical presentation, comorbidities, and results of other tests, including assessment of the right ventricle. Thus, the optimal approach ranges from no further investigation to right heart catheterization and, in some cases, referral to a pulmonary hypertension center.
THE TWO MEASUREMENTS COMPARED
Although it raises concern, the finding of high systolic pulmonary artery pressure is not enough to diagnose pulmonary hypertension. In fact, several other conditions are associated with high systolic pulmonary artery pressure on echocardiography (Table 1). The diagnosis must be confirmed with right heart catheterization.1
Echocardiography provides an estimate of the systolic pulmonary artery pressure that is calculated from other values, whereas right heart catheterization gives a direct measurement of the mean pulmonary artery pressure, which is necessary for diagnosing pulmonary hypertension. The two values are correlated, but the differences are noteworthy.
WHAT IS PULMONARY HYPERTENSION?
Pulmonary hypertension is defined by a resting mean pulmonary artery pressure 25 mm Hg or greater during right heart catheterization.1 The large number of conditions associated with pulmonary hypertension can be divided into five groups2:
- Group 1, pulmonary artery hypertension
- Group 2, pulmonary hypertension associated with left heart disease
- Group 3, pulmonary hypertension due to chronic lung disease or hypoxia
- Group 4, chronic thromboembolic pulmonary hypertension
- Group 5, pulmonary hypertension due to unclear multifactorial mechanisms.2
Pulmonary artery hypertension (group 1) is a syndrome characterized by a restricted flow of small pulmonary arteries that can be idiopathic, heritable, or induced by anorexigens, connective tissue disease, congenital heart disease, portal hypertension, human immunodeficiency virus (HIV), or schistosomiasis.2,3 In spite of significant advances in therapy in the last 3 decades, pulmonary artery hypertension continues to lead to right heart failure and death,4 and the diagnosis has adverse prognostic implications. Therefore, it is essential to be attentive when reviewing the echocardiogram, since an elevated systolic pulmonary artery pressure may be an important clue to pulmonary hypertension.
ESTIMATED PRESSURE: HOW HIGH IS TOO HIGH?
There is no consensus on the optimal cutoff of echocardiographic systolic pulmonary artery pressure to trigger a further evaluation for pulmonary hypertension.
A retrospective evaluation of nearly 16,000 normal echocardiograms found that the 95% upper limit for systolic pulmonary artery pressure was 37 mm Hg.5
European guidelines6 propose that pulmonary hypertension is unlikely if the estimated systolic pulmonary artery pressure is 36 mm Hg or lower, possible if it is 37 to 50 mm Hg, and likely if it is higher than 50 mm Hg.6
The 2009 consensus document of the American College of Cardiology Foundation and American Heart Association3 recommends a systolic pulmonary artery pressure greater than 40 mm Hg as the threshold to suggest further evaluation in a patient with unexplained dyspnea.
Converting the systolic pulmonary artery pressure to the mean pressure
Although not validated to use with echocardiography, the most accurate estimate of mean pulmonary artery pressure was shown in one study7 to be obtained with the equation:
0.61 × systolic pulmonary artery pressure
+ 2 mm Hg
Using this formula, a systolic pulmonary artery pressure of 37 mm Hg would correspond to a mean pulmonary artery pressure of 24.6 mm Hg. A systolic pulmonary artery pressure of 40 mm Hg would correspond to a mean pulmonary artery pressure of 26.4 mm Hg.
Estimated systolic pulmonary artery pressure depends on several variables
Systolic pulmonary artery pressure is estimated using the simplified Bernoulli equation8:
4 × tricuspid regurgitation jet velocity2 (m/s)
+ right atrial pressure (mm Hg)
Tricuspid regurgitation is present in over 75% of the normal population. The regurgitation velocity across the tricuspid valve must be measured to estimate the pressure gradient between the right ventricle and the right atrium. The right atrial pressure is estimated from the diameter of the inferior vena cava and the degree of inspiratory collapse with the sniff test. As the right atrial pressure increases, the inferior vena cava dilates and inspiratory collapse decreases.8 If there is no gradient across the right ventricular outflow tract or pulmonary valve, the right ventricular systolic pressure is equal to the systolic pulmonary artery pressure.
Since tricuspid regurgitation velocity is squared and then multiplied by 4, small deviations of this measurement lead to markedly different systolic pulmonary artery pressure values. To avoid this problem, the tricuspid regurgitation velocity needs to be looked at in multiple echocardiographic views to find the best alignment with the flow and an adequate envelope.
Many causes of high estimated systolic pulmonary artery pressure
Table 1 shows conditions associated with a high estimated systolic pulmonary artery pressure. Echocardiographic limitations, constitutional factors, and high cardiac output states can lead to an apparent elevation in systolic pulmonary artery pressure, which is not confirmed later during right heart catheterization.
Systolic pulmonary artery pressure increases with age and body mass index as a result of worsening left ventricular diastolic dysfunction.8 In fact, an estimated pressure greater than 40 mm Hg is found5 in 6% of people over age 50 and in 5% of people with a body mass index greater than 30 kg/m2. It can also be high in conditions in which there is an increase in cardiac output, such as pregnancy, anemia (sickle cell disease, thalassemia), cirrhosis, and arteriovenous fistula.
The estimated systolic value often differs from the measured value
Studies have compared the systolic pulmonary artery pressure measured during right heart catheterization with the estimated value on echocardiography.9,10 These studies noted a reasonable degree of agreement between the tests but a substantial variability.
Both underestimation and overestimation of the systolic pulmonary artery pressure by echocardiography were common, with 95% limits of agreement ranging from minus 40 mm Hg to plus 40 mm Hg.9,10 A difference of plus or minus 10 mm Hg in systolic pulmonary artery pressure between echocardiography and catheterization was observed in 48% to 51% of patients with pulmonary hypertension, particularly in those with higher systolic pulmonary artery pressure.9,10
An important reason for overestimation of systolic pulmonary artery pressure is the inaccurate estimation of the right atrial pressure by echocardiography.9,10 Indeed, this factor may account for half of the cases in which the systolic pulmonary artery pressure is overestimated.10 Although the traditional methods to estimate the right atrial pressure have been revisited,8,11 this estimation is less reliable for intermediate pressure values, for patients on mechanical ventilation, and for young athletes.8
Other explanations for the variability between measured and estimated systolic pulmonary artery pressure include suboptimal alignment between the Doppler beam and the regurgitant jet, severe tricuspid regurgitation, arrhythmias, and limitations inherent to the simplified Bernoulli equation.12 The estimated value is particularly inaccurate in patients with advanced lung disease, possibly owing to lung hyperinflation and alteration in the thoracic cavity and position of the heart—all factors that limit visualization and measurement of the tricuspid regurgitant jet.13
OTHER SIGNS OF PULMONARY HYPERTENSION ON ECHOCARDIOGRAPHY
Echocardiography provides information that is useful in assessing the accuracy of the estimated systolic pulmonary artery pressure, particularly right ventricular size and function.
As pulmonary hypertension progresses, the right ventricle dilates, and its function is compromised. Therefore, it is important to determine the right ventricular size and function by using objective echocardiographic findings such as right ventricular diameters (basal, mid, apical) and area, right ventricular fractional area change, tricuspid annular plane systolic excursion, myocardial performance index, and the pulsed tissue Doppler tricuspid annular peak systolic excursion velocity.8
Other echocardiographic features that suggest pulmonary hypertension include a dilated right atrial area, flattening of the interventricular septum, notching of the right ventricular outflow tract flow, and dilation of the main pulmonary artery. Interestingly, left ventricular diastolic dysfunction of the impaired relaxation type (grade I) is commonly observed in pulmonary hypertension14; however, more advanced degrees of diastolic dysfunction, ie, pseudonormalization (grade II) or restrictive left ventricular filling (grade III),15 particularly when associated with a left atrial enlargement, suggest pulmonary hypertension associated with left heart disease and not pulmonary artery hypertension.
WHAT TO DO IF ECHOCARDIOGRAPHY INDICATES PULMONARY HYPERTENSION
An algorithm showing the approach to an elevated systolic pulmonary artery pressure on echocardiography is presented in Figure 1.
In the appropriate clinical setting, if the systolic pulmonary artery pressure is 40 mm Hg or greater or if other echocardiographic variables suggest pulmonary hypertension, our practice is to proceed with right heart catheterization.
Clinical variables that suggest pulmonary hypertension include progressive dyspnea, chest pain, presyncope-syncope, lower extremity edema, hepatomegaly, jugular vein distention, hepatojugular reflux, sternal heave, loud second heart sound (P2), murmur of tricuspid or pulmonary regurgitation, and right ventricular third heart sound.16 These are of particular interest when associated with conditions known to cause pulmonary hypertension,2such as connective tissue disease, portal hypertension, congenital heart disease, HIV infection, and certain drugs and toxins.
Other tests that raise suspicion of pulmonary hypertension are an electrocardiogram suggesting a dilated right atrium or ventricle, an elevated brain natriuretic peptide level, a low carbon monoxide diffusing capacity on pulmonary function testing, and an enlarged pulmonary artery diameter on imaging.
Given the high prevalence of pulmonary hypertension, the Fifth World Symposium on Pulmonary Hypertension recommended first considering heart or parenchymal lung disease when an echocardiogram suggests pulmonary hypertension.6 If there are signs of severe pulmonary hypertension or right ventricular dysfunction, referral to a center specializing in pulmonary hypertension is recommended. Referral is also appropriate when there is no major heart or lung disease and the echocardiogram shows an elevated systolic pulmonary artery pressure, particularly when the clinical presentation or results of other testing suggest pulmonary hypertension.
TAKE-HOME POINTS
In the appropriate context, a high systolic pulmonary artery pressure on echocardiography suggests pulmonary hypertension, but right heart catheterization is needed to confirm the diagnosis. Estimating the systolic pulmonary artery pressure with echocardiography has limitations, including false-positive results, predominantly when the pretest probability of pulmonary hypertension is low.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D42–D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D34–D41.
- McLaughlin VV, Archer SL, Badesch DB, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009; 53:1573–1619.
- Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013; 188:365–369.
- McQuillan BM, Picard MH, Leavitt M, Weyman AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation 2001; 104:2797–2802.
- Galiè N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30:2493–2537.
- Chemla D, Castelain V, Provencher S, Humbert M, Simonneau G, Herve P. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest 2009; 135:760–768.
- Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 2010; 23:685–713.
- Rich JD, Shah SJ, Swamy RS, Kamp A, Rich S. Inaccuracy of Doppler echocardiographic estimates of pulmonary artery pressures in patients with pulmonary hypertension: implications for clinical practice. Chest 2011; 139:988–993.
- Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med 2009; 179:615–621.
- Brennan JM, Blair JE, Goonewardena S, et al. Reappraisal of the use of inferior vena cava for estimating right atrial pressure. J Am Soc Echocardiogr 2007; 20:857–861.
- Giardini A, Tacy TA. Non-invasive estimation of pressure gradients in regurgitant jets: an overdue consideration. Eur J Echocardiogr 2008; 9:578–584.
- Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003; 167:735–740.
- Tonelli AR, Plana JC, Heresi GA, Dweik RA. Prevalence and prognostic value of left ventricular diastolic dysfunction in idiopathic and heritable pulmonary arterial hypertension. Chest 2012; 141:1457–1465.
- Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009; 22:107–133.
- Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43(suppl S):40S–47S.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D42–D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(suppl D):D34–D41.
- McLaughlin VV, Archer SL, Badesch DB, et al; American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 2009; 53:1573–1619.
- Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013; 188:365–369.
- McQuillan BM, Picard MH, Leavitt M, Weyman AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation 2001; 104:2797–2802.
- Galiè N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30:2493–2537.
- Chemla D, Castelain V, Provencher S, Humbert M, Simonneau G, Herve P. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest 2009; 135:760–768.
- Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 2010; 23:685–713.
- Rich JD, Shah SJ, Swamy RS, Kamp A, Rich S. Inaccuracy of Doppler echocardiographic estimates of pulmonary artery pressures in patients with pulmonary hypertension: implications for clinical practice. Chest 2011; 139:988–993.
- Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med 2009; 179:615–621.
- Brennan JM, Blair JE, Goonewardena S, et al. Reappraisal of the use of inferior vena cava for estimating right atrial pressure. J Am Soc Echocardiogr 2007; 20:857–861.
- Giardini A, Tacy TA. Non-invasive estimation of pressure gradients in regurgitant jets: an overdue consideration. Eur J Echocardiogr 2008; 9:578–584.
- Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003; 167:735–740.
- Tonelli AR, Plana JC, Heresi GA, Dweik RA. Prevalence and prognostic value of left ventricular diastolic dysfunction in idiopathic and heritable pulmonary arterial hypertension. Chest 2012; 141:1457–1465.
- Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009; 22:107–133.
- Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43(suppl S):40S–47S.
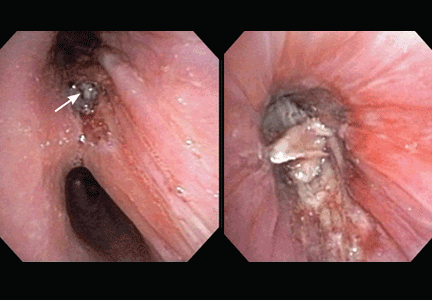
A 19-year-old man with progressive lung infiltrates
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
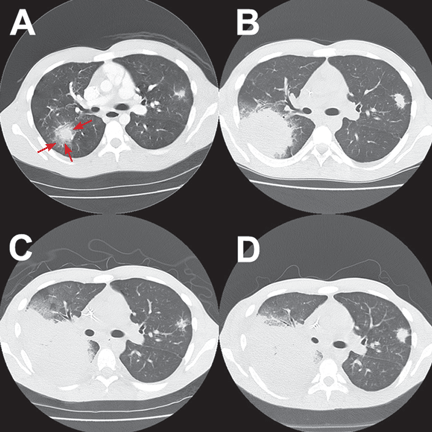
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
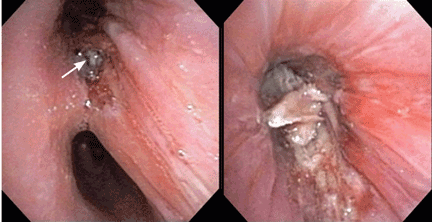
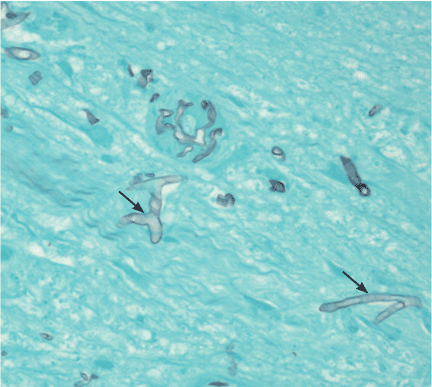
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.