User login
Pediatric Dermatology Consult - July 2018
Frequently misdiagnosed, streptococcal intertrigo more commonly affects infants and toddlers but is rarely reported, especially compared with other Streptococcus pyogenes infections, including impetigo, erysipelas, and cellulitis.1
Intertrigo, meaning “between” (inter) and “to rub” (terere) in Latin, describes any skin disorder involving two opposing skin surfaces that touch or rub to cause friction.2 The continuous chaffing, coupled with moisture trapped within the skin folds, leads to irritation and maceration, which provides an ideal environment for pathogens to thrive. Thus, frictional dermatitides that arise may become secondarily infected with one or more microorganisms, such as Candida albicans, Staphylococcus aureus, Streptococcus pyogenes, and even organisms less commonly associated with cutaneous infection, such as Proteus mirabilis.3
Streptococcal intertrigo may affect any intertriginous area, but most commonly it affects the folds of the neck; this is likely because of the combination of the deep folds that develop in shorter, infantile necks and the moisture from drool and saliva that pools in the area.5,6 In addition to these cervical folds, other intertriginous areas commonly are affected, including the inguinal, axillary, popliteal, posterior auricular, perianal, and genital folds.
Perianal streptococcal disease may present in a similar manner as streptococcal intertrigo, manifesting as well-demarcated, beefy red plaques in the skin folds around the anus and, in females, frequently perivaginally.7 Unlike streptococcal intertrigo, perianal streptococcal disease is often characterized by pain, pruritus, and fissuring of the involved area.8 It is associated with pharyngeal colonization of group A beta-hemolytic streptococci.7
Diagnosis is straight forward and may be confirmed by a positive streptococcal rapid antigen test of swab specimens of one or more surfaces of affected skin or by culture from a skin swab yielding growth of the organism.1,5 Skin biopsy is not necessary. If the index of suspicion for candida is high, a potassium hydroxide preparation and culture may be performed. Checking serum anti-DNase B antibodies, antistreptolysin O, and pharyngeal cultures is often unrevealing.9 A urinalysis may be performed to assess for poststreptococcal glomerulonephritis if the patient later develops facial or orbital edema, hypertension, hematuria, or lethargy.9
Treatment consists of systemic antistreptococcal therapy; oral amoxicillin and penicillin frequently have been used.9 Moisture in the area should be reduced with application of absorptive powders and physical barriers, such as zinc oxide, after gentle cleansing of the area.5
Other diagnoses to consider when evaluating dermatitides affecting skin folds include: other infectious causes, which may be ruled out by fungal or bacterial culture; inverse psoriasis, which will frequently demonstrate scale; atopic dermatitis, which will be pruritic with history of atopy; irritant or contact dermatitis, which will often have correlating clinical history; seborrheic dermatitis, which will often involve greasiness and scale; and less commonly, acrodermatitis enteropathica, which will be accompanied by diarrhea and hair loss.2,9 Scabies also may be on the differential if the patient endorses severe pruritus with close contacts with similar symptoms.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university. They had no conflicts of interest or disclosures to report.
References
1. Pediatr Dermatol. 2014 Mar-Apr;31(2):e71-2.
2. Clin Dermatol. 2011 Mar-Apr;29(2):173-9.
3. Pediatrics. 2003 Dec;112(6 pt 1):1427-9.
4. BMJ Case Rep. 2018 Mar 20. doi: 10.1136/bcr-2018-224179.
5. Pediatr Infect Dis J. 2012 Aug;31(8):872-3.
6. J Pediatr. 2015 May;166(5):1318.
7. J Pediatr. 2015 Sep;167(3):687-93.e1-2.
8. Pediatrics in Review. 1991;12(8):248-55.
9. J Pediatr. 2017 May;184:230-1.e1.
Frequently misdiagnosed, streptococcal intertrigo more commonly affects infants and toddlers but is rarely reported, especially compared with other Streptococcus pyogenes infections, including impetigo, erysipelas, and cellulitis.1
Intertrigo, meaning “between” (inter) and “to rub” (terere) in Latin, describes any skin disorder involving two opposing skin surfaces that touch or rub to cause friction.2 The continuous chaffing, coupled with moisture trapped within the skin folds, leads to irritation and maceration, which provides an ideal environment for pathogens to thrive. Thus, frictional dermatitides that arise may become secondarily infected with one or more microorganisms, such as Candida albicans, Staphylococcus aureus, Streptococcus pyogenes, and even organisms less commonly associated with cutaneous infection, such as Proteus mirabilis.3
Streptococcal intertrigo may affect any intertriginous area, but most commonly it affects the folds of the neck; this is likely because of the combination of the deep folds that develop in shorter, infantile necks and the moisture from drool and saliva that pools in the area.5,6 In addition to these cervical folds, other intertriginous areas commonly are affected, including the inguinal, axillary, popliteal, posterior auricular, perianal, and genital folds.
Perianal streptococcal disease may present in a similar manner as streptococcal intertrigo, manifesting as well-demarcated, beefy red plaques in the skin folds around the anus and, in females, frequently perivaginally.7 Unlike streptococcal intertrigo, perianal streptococcal disease is often characterized by pain, pruritus, and fissuring of the involved area.8 It is associated with pharyngeal colonization of group A beta-hemolytic streptococci.7
Diagnosis is straight forward and may be confirmed by a positive streptococcal rapid antigen test of swab specimens of one or more surfaces of affected skin or by culture from a skin swab yielding growth of the organism.1,5 Skin biopsy is not necessary. If the index of suspicion for candida is high, a potassium hydroxide preparation and culture may be performed. Checking serum anti-DNase B antibodies, antistreptolysin O, and pharyngeal cultures is often unrevealing.9 A urinalysis may be performed to assess for poststreptococcal glomerulonephritis if the patient later develops facial or orbital edema, hypertension, hematuria, or lethargy.9
Treatment consists of systemic antistreptococcal therapy; oral amoxicillin and penicillin frequently have been used.9 Moisture in the area should be reduced with application of absorptive powders and physical barriers, such as zinc oxide, after gentle cleansing of the area.5
Other diagnoses to consider when evaluating dermatitides affecting skin folds include: other infectious causes, which may be ruled out by fungal or bacterial culture; inverse psoriasis, which will frequently demonstrate scale; atopic dermatitis, which will be pruritic with history of atopy; irritant or contact dermatitis, which will often have correlating clinical history; seborrheic dermatitis, which will often involve greasiness and scale; and less commonly, acrodermatitis enteropathica, which will be accompanied by diarrhea and hair loss.2,9 Scabies also may be on the differential if the patient endorses severe pruritus with close contacts with similar symptoms.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university. They had no conflicts of interest or disclosures to report.
References
1. Pediatr Dermatol. 2014 Mar-Apr;31(2):e71-2.
2. Clin Dermatol. 2011 Mar-Apr;29(2):173-9.
3. Pediatrics. 2003 Dec;112(6 pt 1):1427-9.
4. BMJ Case Rep. 2018 Mar 20. doi: 10.1136/bcr-2018-224179.
5. Pediatr Infect Dis J. 2012 Aug;31(8):872-3.
6. J Pediatr. 2015 May;166(5):1318.
7. J Pediatr. 2015 Sep;167(3):687-93.e1-2.
8. Pediatrics in Review. 1991;12(8):248-55.
9. J Pediatr. 2017 May;184:230-1.e1.
Frequently misdiagnosed, streptococcal intertrigo more commonly affects infants and toddlers but is rarely reported, especially compared with other Streptococcus pyogenes infections, including impetigo, erysipelas, and cellulitis.1
Intertrigo, meaning “between” (inter) and “to rub” (terere) in Latin, describes any skin disorder involving two opposing skin surfaces that touch or rub to cause friction.2 The continuous chaffing, coupled with moisture trapped within the skin folds, leads to irritation and maceration, which provides an ideal environment for pathogens to thrive. Thus, frictional dermatitides that arise may become secondarily infected with one or more microorganisms, such as Candida albicans, Staphylococcus aureus, Streptococcus pyogenes, and even organisms less commonly associated with cutaneous infection, such as Proteus mirabilis.3
Streptococcal intertrigo may affect any intertriginous area, but most commonly it affects the folds of the neck; this is likely because of the combination of the deep folds that develop in shorter, infantile necks and the moisture from drool and saliva that pools in the area.5,6 In addition to these cervical folds, other intertriginous areas commonly are affected, including the inguinal, axillary, popliteal, posterior auricular, perianal, and genital folds.
Perianal streptococcal disease may present in a similar manner as streptococcal intertrigo, manifesting as well-demarcated, beefy red plaques in the skin folds around the anus and, in females, frequently perivaginally.7 Unlike streptococcal intertrigo, perianal streptococcal disease is often characterized by pain, pruritus, and fissuring of the involved area.8 It is associated with pharyngeal colonization of group A beta-hemolytic streptococci.7
Diagnosis is straight forward and may be confirmed by a positive streptococcal rapid antigen test of swab specimens of one or more surfaces of affected skin or by culture from a skin swab yielding growth of the organism.1,5 Skin biopsy is not necessary. If the index of suspicion for candida is high, a potassium hydroxide preparation and culture may be performed. Checking serum anti-DNase B antibodies, antistreptolysin O, and pharyngeal cultures is often unrevealing.9 A urinalysis may be performed to assess for poststreptococcal glomerulonephritis if the patient later develops facial or orbital edema, hypertension, hematuria, or lethargy.9
Treatment consists of systemic antistreptococcal therapy; oral amoxicillin and penicillin frequently have been used.9 Moisture in the area should be reduced with application of absorptive powders and physical barriers, such as zinc oxide, after gentle cleansing of the area.5
Other diagnoses to consider when evaluating dermatitides affecting skin folds include: other infectious causes, which may be ruled out by fungal or bacterial culture; inverse psoriasis, which will frequently demonstrate scale; atopic dermatitis, which will be pruritic with history of atopy; irritant or contact dermatitis, which will often have correlating clinical history; seborrheic dermatitis, which will often involve greasiness and scale; and less commonly, acrodermatitis enteropathica, which will be accompanied by diarrhea and hair loss.2,9 Scabies also may be on the differential if the patient endorses severe pruritus with close contacts with similar symptoms.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university. They had no conflicts of interest or disclosures to report.
References
1. Pediatr Dermatol. 2014 Mar-Apr;31(2):e71-2.
2. Clin Dermatol. 2011 Mar-Apr;29(2):173-9.
3. Pediatrics. 2003 Dec;112(6 pt 1):1427-9.
4. BMJ Case Rep. 2018 Mar 20. doi: 10.1136/bcr-2018-224179.
5. Pediatr Infect Dis J. 2012 Aug;31(8):872-3.
6. J Pediatr. 2015 May;166(5):1318.
7. J Pediatr. 2015 Sep;167(3):687-93.e1-2.
8. Pediatrics in Review. 1991;12(8):248-55.
9. J Pediatr. 2017 May;184:230-1.e1.
An 8-week-old male with a history of cradle cap presented for a second evaluation of an erythematous rash on the neck that started 1.5 weeks before, and it had since worsened. The parents note that their infant has been more irritable, but they otherwise deny any fever, diarrhea, constipation, or decrease in oral intake.
The patient’s first evaluation had been 3 days prior; nystatin cream was prescribed, and the parents applied it twice a day but without improvement to the rash. The patient also had a rash behind the ears bilaterally, which was treated with hydrocortisone 2.5% ointment with some improvement
On physical exam, the central neck is covered by a bright, beefy red, erythematous plaque with distinct borders and strong odor. There is faint scale and superficial desquamation between the skin folds. There are no surrounding papules or pustules. The patient’s chin is moist with drool. In the postauricular skin folds bilaterally, there are fainter but still erythematous plaques with mild scale.
Pediatric Dermatology Consult - March 2018
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.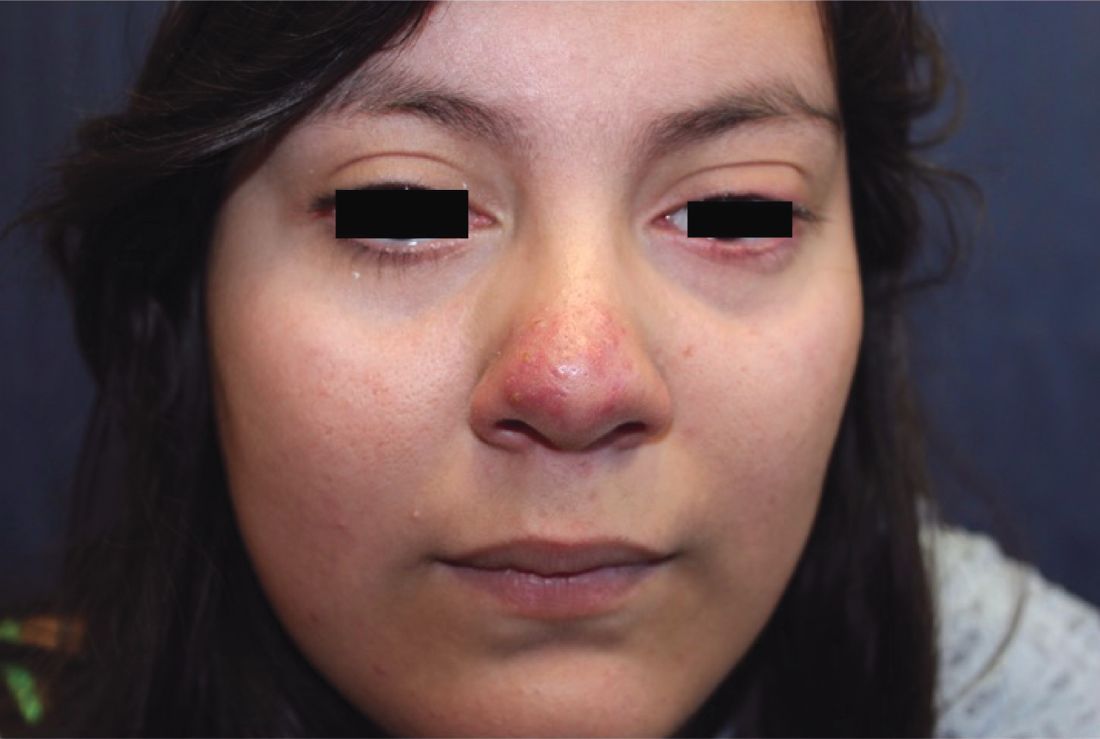
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2
Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2
Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2
Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2
Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2
Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2
Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
A 16-year-old girl presented with a 6-month history of an erythematous eruption of small papules and pustules around the cheeks and nose. She states the erythema had started first, with periods of feeling flushed that became worse with sun exposure. She saw her primary care physician who prescribed topical steroids. After using the steroids, the rash became worse, and she developed papules and pustules.
Pediatric Dermatology Consult - January 2018
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
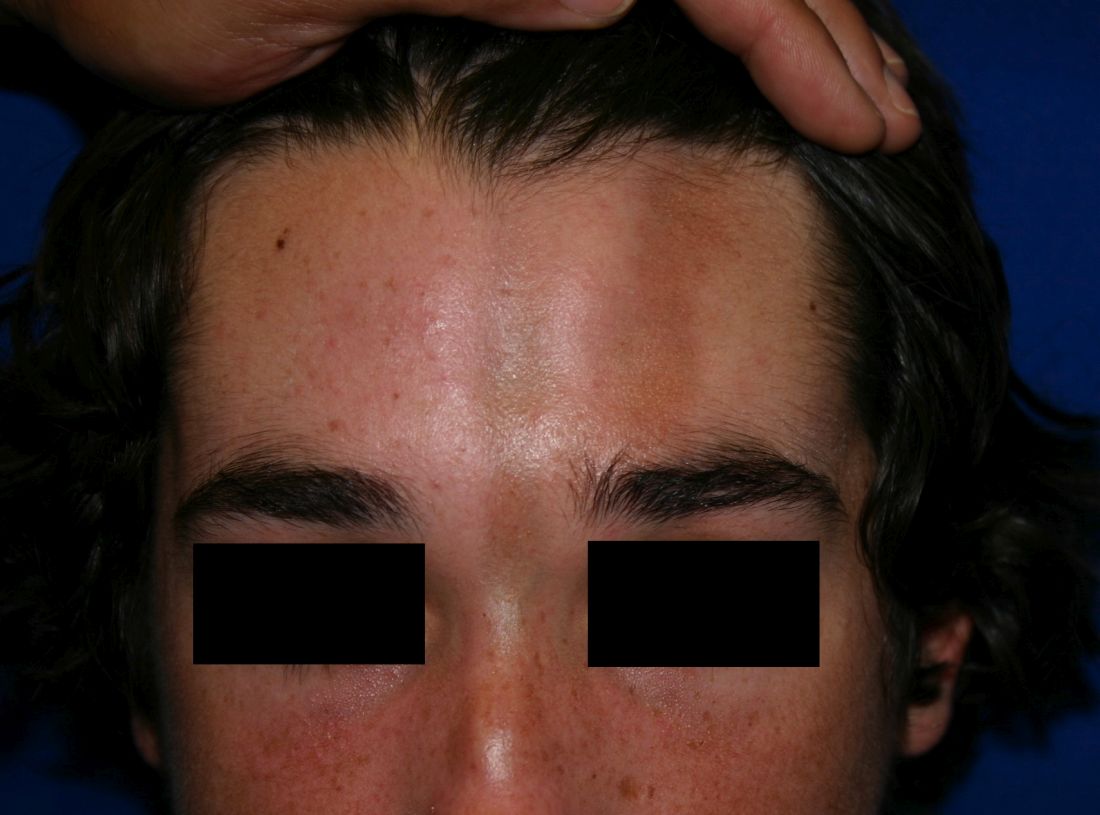
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
A 14-year-old patient presents to a dermatology clinic for a depression on his forehead, which has been there for about 2 years. A few years ago, he used to have a pruritic pink lesion on the forehead where the depression is now. He denies any symptoms.
Confluent and reticulated papillomatosis
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.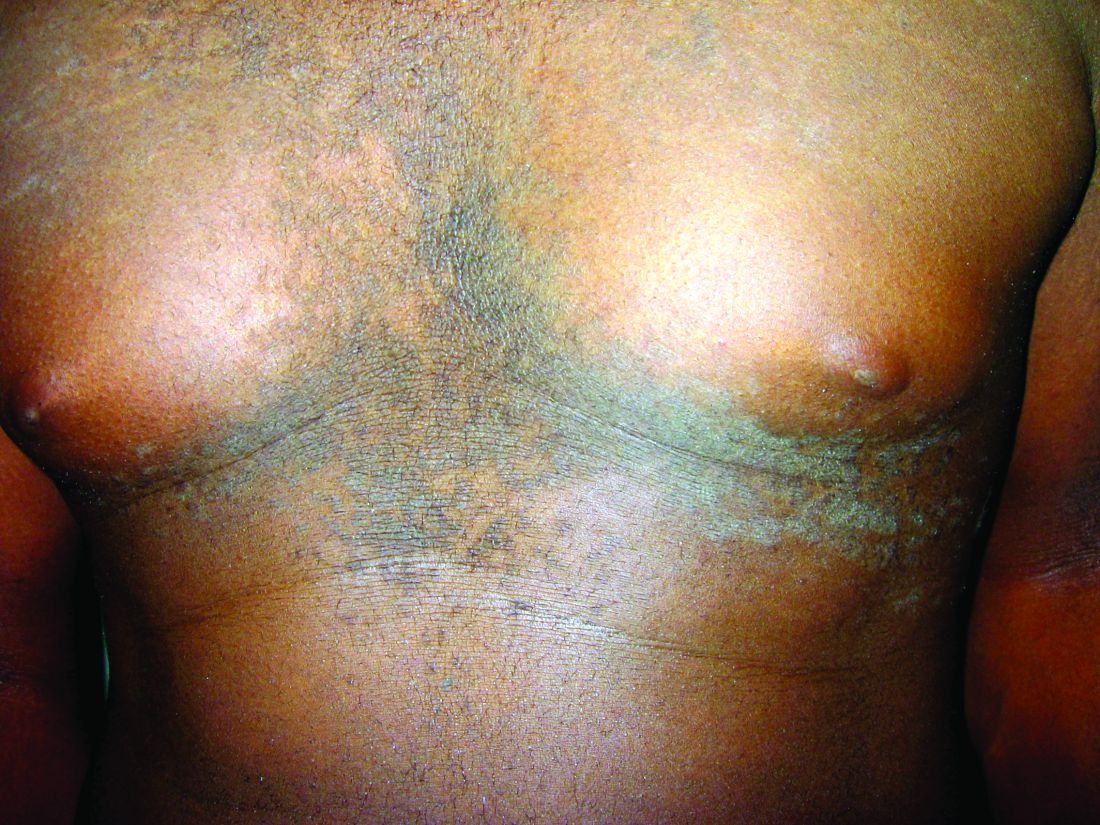
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
A 17-year-old male presents to the dermatology clinic for brown lesions on his central chest and back that have been present for about a year. The brown areas gradually have become scaly over time. They are asymptomatic. His pediatrician had given him hydrocortisone ointment to apply to the lesions, but there was no improvement. Review of systems was otherwise negative.