User login
Breakdown
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
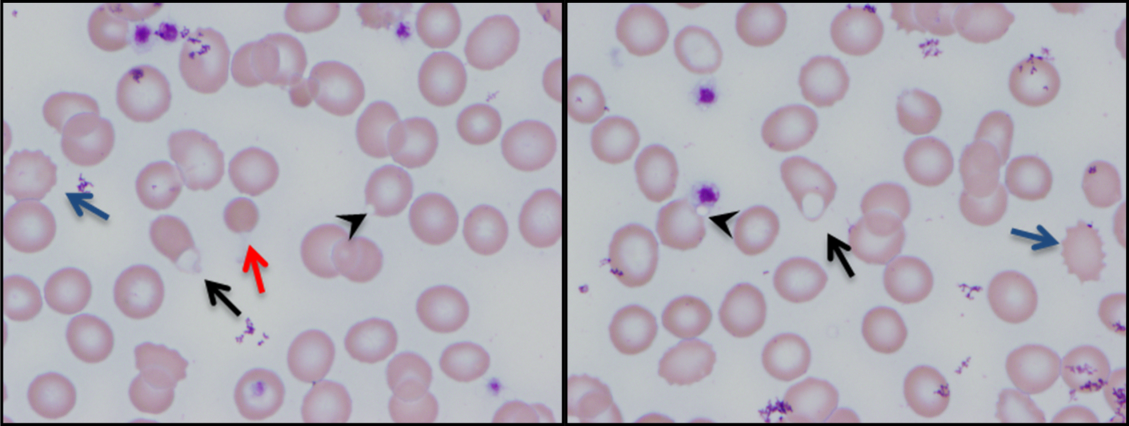
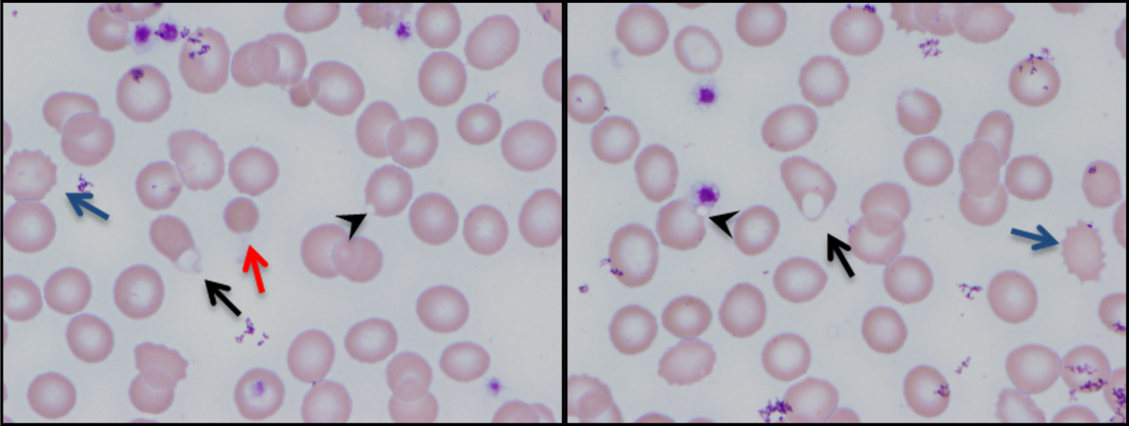
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
Not a Textbook Case
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
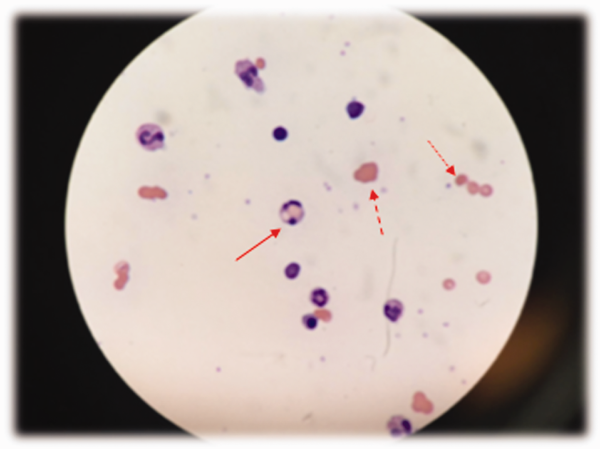
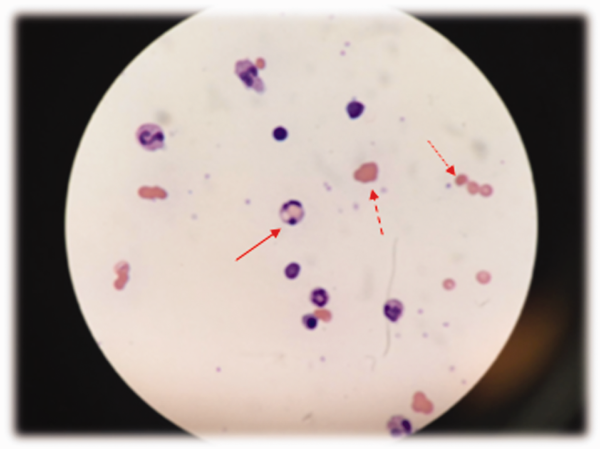
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
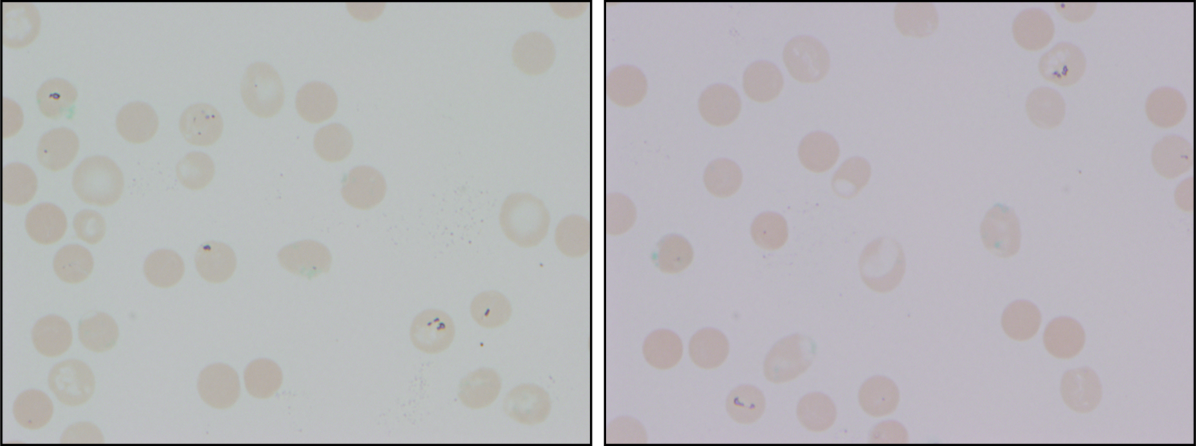
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.