User login
Management of hyponatremia: Providing treatment and avoiding harm
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT

The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?

Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
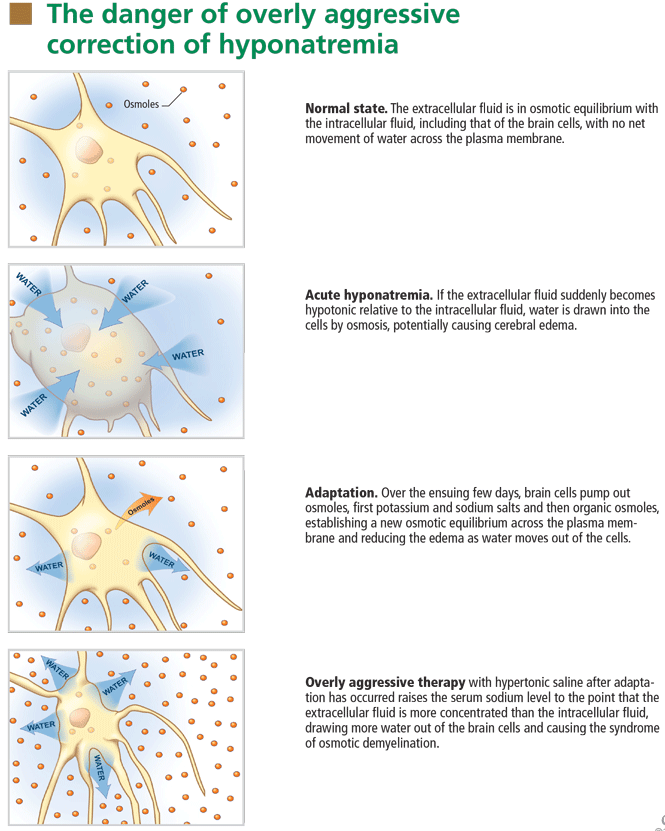
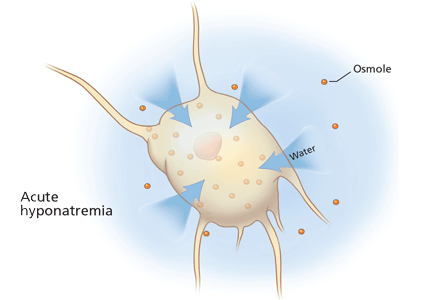
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment

Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia

TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14

An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3 Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
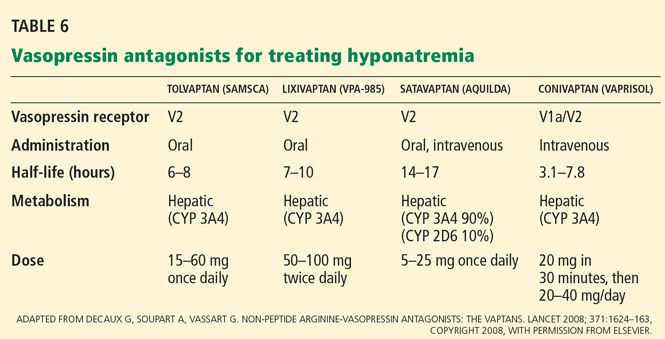
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
- Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
- In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
- Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
- There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
- Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
- Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet 1981; 2:26–31.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med 2007; 120(suppl 1):S1–S21.
- Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med 2004; 71:639–650.
- Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med 1989; 86:315–318.
- Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant 2003; 18:2486–2491.
- Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med 2007; 74:377–383.
- Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol 1987; 252:F661–F669.
- Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol 1997; 8:1599–1607.
- Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int 2006; 69:1291–1293.
- Ellis SJ. Severe hyponatraemia: complications and treatment. QJM 1995; 88:905–909.
- Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol 1994; 4:1522–1530.
- Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol 2008; 3:331–336.
- Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int 2009; 76:587–589.
- Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol 2004; 8:12–16.
- Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med 2000; 342:1581–1589.
- Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol 2007; 2:1110–1117.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med 2007; 356:2064–2072.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med 2006; 119:71.e1–e8.
- Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol 2010; 5:275–280.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med 1987; 83:905–988.
- Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM 2005; 98:529–540.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol 2008; 19:1076–1078.
- Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med 1977; 87:195–197.
- Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists? J Am Soc Nephrol 2008; 19:1054–1058.
- Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet 2008; 371:1624–1632.
- Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 2006; 355:2099–2112.
- Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007; 297:1319–1331.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol 2010; 21:705–712.
- Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if? J Am Soc Nephrol 2010; 21:552–555.
- Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int 2006; 69:2124–2130.
- Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol 2007; 2:151–161.
- Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med 2005; 353:427–428.
- Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med 2005; 15:208–213.
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT
The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?
Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment
Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia
TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14
An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3 Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
- Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
- In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
- Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
- There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
- Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is one of the most frequently encountered electrolyte disorders. In 1981, Flear et al1 reported that 15% of their hospitalized patients had plasma sodium concentrations lower than 134 mmol/L, the cutoff they were using at that time.
Hyponatremia is sometimes merely a laboratory artifact or a result of improper blood collection. If real, it can be due to excessive water intake or, most often, the inability of the kidney to excrete water coupled with continued water intake. Patients with significant underlying cardiac, hepatic, or renal dysfunction are at greatest risk of developing hyponatremia, secondary to the nonosmotic release of antidiuretic hormone (ADH). Others at risk include postoperative patients (especially menstruating women), older patients on thiazide diuretics, patients with malignant or psychiatric illness, and endurance athletes.
In this article, we review the treatment of acute and chronic hyponatremia, emphasizing the importance of basing the therapy on the severity of symptoms and taking care not to raise the serum sodium level too rapidly, which can cause neurologic dysfunction.
Guidelines for managing hyponatremia2 are based mostly on retrospective studies and expert opinion, since few prospective studies have been done. Despite the paucity of evidence-based recommendations, we will attempt to incorporate findings from important human and animal studies and consensus guidelines from expert panels. We will focus initially on the critical diagnostic considerations necessary to initiate treatment.
SYMPTOMATIC VS ASYMPTOMATIC
Subsequent sections will address therapeutic approaches in two clinical settings:
Symptomatic hyponatremia, ie, with severe signs or symptoms of cerebral edema—a medical emergency; and
Asymptomatic hyponatremia, ie, without serious signs or symptoms of cerebral edema.
KEY DIAGNOSTIC STEPS WHEN STARTING TREATMENT
The clinical and laboratory evaluations form the foundation of a proper approach to any patient with hyponatremia. The rationale behind making several important diagnostic distinctions will be discussed here briefly and then expanded on in the remaining text. The reader is referred to another review on the diagnostic evaluation of hyponatremia.3
Confirm that the patient truly has hypo-osmolar hyponatremia
The serum osmolality should be measured to confirm that it is low (ie, < 275 mOsm/kg). In addition, the arterial serum sodium concentration can be measured using a blood gas device if pseudohyponatremia (see below) is suspected. This method uses direct potentiometry and bypasses the dilutional step in the processing of venous samples.4
Rationale. The clinical consequences of hyponatremia are due to water moving from hypo-osmolar extracellular fluid into the relatively hyperosmolar interior of the cell. This water movement can cause progressive cerebral edema, resulting in a spectrum of signs and symptoms from headache and ataxia to seizures and coma. But significant fluid shifts and cerebral edema occur only if the extracellular fluid is hypo-osmolar relative to the intracellular fluid.
In fact, hyponatremia can occur in several situations in which the extracellular fluid is not hypo-osmolar. An increase in effective plasma osmoles (substances in the extracellular fluid that do not readily move across the plasma membrane) can cause water to move out of cells, resulting in translocational hyponatremia. This may be seen in hyperglycemia or when mannitol or contrast dye has been given. In these situations, the plasma is either isotonic or even hypertonic to the intracellular fluid, resulting in no movement of water into the cells and therefore no clinical consequences relating to the hyponatremia. Importantly, no therapy is required for the hyponatremia.
Other situations in which hyponatremia is present but not associated with true hypotonicity include states of excess protein or lipid in the blood (pseudohyponatremia). Also, if an infusion of hypotonic fluid is running, clinicians must be sure that blood samples are not drawn proximally in the same vein.
Are there significant signs or symptoms of cerebral edema?
Patients need to be assessed quickly because those with serious neurologic signs or symptoms thought to be related to hyponatremia require urgent treatment with hypertonic saline to increase the serum sodium concentration, regardless of the underlying volume status, the cause of hyponatremia, or the time of onset.
Determine the duration of hyponatremia
One should try to ascertain when the hyponatremia started, as its duration is important in determining the proper pace of correction.
At the onset of hyponatremia, water moves from the extracellular fluid into cells, pulled in by osmosis. The brain can decrease the net amount of water entering into the neurons (and thus regulate its volume) by increasing the flow of water from the interstitium into the cerebrospinal fluid via increased interstitial hydraulic pressure.7
Over the next several days, inorganic solutes (eg, potassium and sodium salts) and various organic solutes are transported out of the cells. In patients in whom this process has had time to occur, treatment of hyponatremia with hypertonic fluids raises the plasma osmolality faster than the cells can recapture the previously transported osmoles. In this situation, overly rapid correction can cause excessive loss of intracellular water, resulting in cell shrinkage and osmotic demyelination syndrome. Osmotic demyelination usually presents during treatment of hyponatremia after an initial improvement in mental status, with worsening neurologic function and various neurologic signs, including paresis and ultimately even death.6
In patients with acute-onset hyponatremia (ie, with onset within the past 48 hours), in whom the above cerebral adaptations have not had time to occur completely, rapid correction is unlikely to result in osmotic demyelination.
In view of the serious risk of osmotic demyelination, if the timing of development of hyponatremia cannot be determined, one should assume it is chronic (> 48 hours) and avoid rapid overcorrection (see discussion below on the rate of correction).
On the other hand, patients who have severe neurologic signs or symptoms initially need their serum sodium increased urgently to safer levels, regardless of the timing of onset (see below for suggested approach). Subsequent treatment of hyponatremia—after the serum sodium level has been raised enough to reverse neurologic symptoms—will be influenced by the duration of the hyponatremia, with careful avoidance of overly rapid correction, especially in patients with chronic hyponatremia.
Assess the patient’s volume status to determine the proper initial treatment
Check urine osmolality to assess for hyponatremic states in which urinary dilution is intact
Measuring urine osmolality is useful in ascertaining whether hyponatremic patients are making appropriately dilute urine (< 100 mOsm/kg). If they are, the cause of the hyponatremia may be excessive water intake, a reset osmostat, or low solute intake. In addition, patients with hypovolemic hyponatremia may have appropriately dilute urine soon after treatment with isotonic intravenous fluids.
The serum sodium concentration often returns to normal if the underlying cause is eliminated (eg, if excessive fluid intake is stopped). If there are no serious signs or symptoms, this can usually be accomplished without additional therapy with intravenous fluids or medications, thereby avoiding the risk of overcorrection.
Search for causes of rapidly correctable hyponatremia
TREATING HYPONATREMIC PATIENTS WITH SERIOUS SIGNS OR SYMPTOMS
Patients with hypo-osmolar hyponatremia and serious signs or symptoms of cerebral edema (lethargy, respiratory depression, seizures) need rapid initial correction of the serum sodium level, as this is a true medical emergency.
Certain patients are at greater risk of developing cerebral edema from hyponatremia (Table 2).
On the other hand, patients with chronic hyponatremia are very unlikely to present with signs or symptoms of cerebral edema. In fact, in a patient with chronic hyponatremia, care must be taken to avoid overcorrection beyond that needed to reverse severe signs and symptoms. In the rare case in which a patient with chronic hyponatremia presents with signs or symptoms of cerebral edema, the hypertonic saline infusion must be stopped as soon as the signs or symptoms have resolved. Further rapid changes in serum sodium must be avoided.
During correction of hyponatremia, some patients are at particularly high risk of osmotic demyelination syndrome secondary to underlying abnormalities in cerebral osmotic regulation. These include patients with alcoholism, malnutrition, hypokalemia, and burns, and elderly women on thiazide diuretics.8 These patients should be monitored vigilantly for overly rapid correction during treatment.
INITIAL TREATMENT: REVERSE CEREBRAL EDEMA WITH 3% SALINE
The goal of the initial, rapid phase of correction is to reverse cerebral edema.
Patients with serious signs or symptoms should receive hypertonic (3%) saline at a rate of about 1 mL/kg/hour for the first several hours.8 Those with concomitant hypervolemia (as in congestive heart failure) or underlying cardiovascular disease should also receive a loop diuretic to aid in free-water excretion and to prevent volume overload from the saline infusion. This regimen usually raises the serum sodium concentration enough (usually by about 1 mmol/L/hour) to decrease cerebral edema and improve symptoms.
In patients having active seizures or showing signs of brain herniation, 3% saline can be given initially at a higher rate of about 2 to 3 mL/kg/hour over the first few hours. An alternative approach is an initial 50-mL bolus of 3% saline and an additional 200 mL given over the subsequent 4 to 6 hours.9
No study has compared the efficacy and safety of these approaches, and clinicians should always monitor extracellular fluid volume status, neurologic status, and serum sodium levels closely in any patient treated with hypertonic saline.
After severe signs and symptoms have resolved, 3% saline is promptly discontinued and appropriate therapy is initiated based on the patient’s volume status and underlying cause of hyponatremia (see discussion below).
NEXT, FIND THE APPROPRIATE RATE OF CORRECTION
After the initial serious signs or symptoms have been addressed with hypertonic saline, management should focus on limiting the rate of correction in patients with chronic hyponatremia or hyponatremia of unknown duration.
Animal studies and retrospective human studies have suggested certain guidelines on the appropriate pace and magnitude of correction during treatment of hyponatremia to avoid osmotic demyelination syndrome.2
Clinicians must not attempt to correct the serum sodium to “normal” values. Although patients with acute hyponatremia may tolerate complete correction, there is little evidence that raising the serum sodium concentration acutely by more than 5 to 8 mmol/L is advantageous. Therefore, correction should be judicious in all patients.
Appropriate rates of correction
A recent expert consensus panel suggested that the serum sodium level be raised by no more than 10 to 12 mmol/L during the first 24 hours of treatment, and by less than 18 mmol/L over 48 hours.2
Patients with chronic hyponatremia and signs or symptoms of cerebral edema should have their sodium level raised at an even slower rate—some recommend less than 10 mmol/L in the first 24 hours.10 Aggressive initial correction at the rate of 1.5 to 2 mmol/hour for the first 3 to 4 hours with 3% saline is indicated until serious symptoms (seizure, obtundation) resolve, but correction beyond 10 to 12 mmol/L in the first 24 hours should be avoided. Hypertonic saline therapy should usually be discontinued well before the serum sodium level has risen this much, to avoid a continuing rise in the sodium level after the infusion has stopped.
While hypertonic saline is being infused, serum sodium levels should be checked every 1 to 2 hours. In a study in 56 patients with severe hyponatremia (serum sodium ≤ 105 mmol/L),11 no neurologic complications were observed in patients with chronic hyponatremia whose serum sodium was corrected by less than 12 mmol/L in 24 hours or by less than 18 mmol/L in 48 hours or in whom the average rate of correction to a serum sodium of 120 mmol/L was less than or equal to 0.55 mmol/L per hour.
If the serum sodium concentration has been overcorrected
Desmopressin is effective in preventing and reversing inadvertent overcorrection of hyponatremia. 12 In one study, desmopressin lowered the sodium concentration by 2 to 9 mmol/L in 14 of 20 patients. None of the patients developed any serious adverse consequences.
In addition, intravenous water (dextrose 5%) can be given alone or in combination with desmopressin to prevent or reverse an excessive increase in serum sodium.13 Such therapy may be considered in patients who continue to excrete hypotonic urine and have already reached a serum sodium concentration that meets or exceeds the recommended rate or magnitude of change.
Formulas for estimating the rate of correction
Various formulas have been devised for estimating the change in serum sodium concentration during treatment of hyponatremia.14
An alternative is the Barsoum-Levine equation, which takes into account ongoing urinary losses. Although it is more cumbersome to calculate, it may be more precise.17
Alternatively, in patients without hypovolemia, the clinician can calculate the amount of urinary excretion of free water required to achieve a specific target serum sodium and then measure hourly urinary water excretion during aquaresis induced by furosemide (Lasix).8 Although more physiologic, this method can be clinically cumbersome, requiring timely handling of urine specimens, accurate recording of urine output, and rapid reporting of laboratory results.
Ultimately, these methods serve only as estimates of the change in serum sodium and do not replace careful monitoring of electrolytes (every 1 to 2 hours during acute therapy) and fastidious assessment for clinical signs or symptoms of osmotic demyelination syndrome.
PATIENTS WITH HYPONATREMIA AND NO SERIOUS SIGNS OR SYMPTOMS
General approach
Hyponatremic patients without serious signs or symptoms of cerebral edema do not require urgent therapy to raise the serum sodium.
Patients with chronic asymptomatic hyponatremia are commonly encountered in clinical practice. As a result of cerebral adaptation, they can appear to have no symptoms despite serum sodium levels as low as 115 to 120 mmol/L. However, even if they have no serious signs or symptoms of cerebral edema, some patients may complain of fatigue, lethargy, nausea, gait abnormalities, and muscle cramps and have evidence of milder forms of neurocognitive impairment.18
In a recent case-control study,18 elderly patients with chronic hyponatremia (mean serum sodium concentration 126 ± 5 mmol/L) were more likely to present to the hospital with falls compared with age-matched controls. Further analysis suggested these patients had marked impairments in gait and attention, which improved in some as the serum sodium increased.
Another recent study19 reported that mild hyponatremia (mean serum sodium concentration 132 mmol/L) was independently associated with the risk of fracture, even after adjustment for known osteoporotic risk factors.
Even when there is no need for acute therapy to raise the serum sodium level, the clinician should scrutinize the medical regimen and available clinical data to rule out reversible causes of water excess. These may include ongoing administration of hypotonic fluids (eg, parenteral nutrition or dextrose 5% to “keep the vein open”) or of medications that cause inappropriate release of ADH (eg, selective serotonin reuptake inhibitors) or that impair water excretion (eg, nonsteroidal anti-inflammatory drugs). The clinician should also search for an underlying diagnosis that predisposes to water retention, such as hypothyroidism, adrenal insufficiency, congestive heart failure, or hepatic or renal failure. If hyponatremia is due to endocrine disease, correction of hypothyroidism or adrenal insufficiency should result in water excretion and improvement in the serum sodium.
If the cause of the hyponatremia is not immediately apparent, treatment can be started on the basis of assessment of the patient’s extracellular fluid volume status using clinical examination and supplementary laboratory data such as the serum uric acid concentration and urinary sodium concentration.3 Table 3 outlines general treatment options for hypoosmolar hyponatremia according to extracellular fluid volume status.
Of note, physical examination alone has poor sensitivity and specificity in assessing extracellular fluid volume status in patients with hyponatremia.20,21 This highlights the importance of spot measurements of urine sodium and serum uric acid and, when appropriate, isotonic intravenous saline challenge to detect occult hypovolemia.
In general, patients with euvolemia are treated with fluid restriction, and patients with hypovolemia are given isotonic saline. Patients with hypervolemia can be difficult to treat, but in general they are prescribed both sodium and fluid restriction. Loop diuretics can be given to promote excretion of water and sodium. Thiazide diuretics are avoided, as they impair urinary dilution and worsen hyponatremia. Attempts should be made to optimize the treatment of the underlying hypervolemic disorder (congestive heart failure, cirrhosis, advanced renal failure). Vasopressin receptor antagonists can also be used in selected cases of hypervolemic or euvolemic hyponatremia (see discussion below).
How to prescribe fluid restriction rationally
Ideally, patients should not ingest any more fluid than they can excrete in urine and insensible losses—otherwise, the serum sodium can continue to decrease.
Water excretion can be estimated from solute intake and urine osmolarity. In theory, a 70-kg person with a typical daily solute intake of about 10 mOsm/kg and intact urinary dilution to a urine osmolarity of 50 mOsm/L can excrete up to 14 L of urine (700 mOsm/50 mOsm/L) per day. However, a patient with the syndrome of inappropriate ADH secretion (SIADH) and a fixed urine osmolality of 700 mOsm/kg would excrete a similar solute load in only 1 L of urine. Thus, any fluid intake in excess of this volume could worsen hyponatremia.
To excrete free water, urinary sodium plus urinary potassium must be less than the serum sodium concentration. In this regard, the necessary degree of fluid restriction can also be estimated made on the basis of the patient’s urinary electrolytes.22
Increased solute intake to augment water excretion
In patients without hypervolemia, solute intake can be increased to augment water excretion. 22 This can be achieved with salt tablets or oral urea. Although urea can be effective, it is not commonly used because it is not available the United States and it has poor gastrointestinal tolerability. In patients whose nutritional intake is limited and who continue to ingest fluids (such as, for example, an elderly patient subsisting on tea and toast) every effort should be made to increase solute intake with high-protein foods or supplements.
DRUGS TO INHIBIT VASOPRESSIN
Unfortunately, patients often do not adhere to these strategies, as fluid restriction and unpalatable salt tablets or urea can become too burdensome. In such instances, pharmacologic inhibition of vasopressin-mediated water reabsorption can be considered using the following agents.
Demeclocycline (Declomycin) and lithium inhibit the kidney’s response to vasopressin. Because lithium may be nephrotoxic and has unwanted effects on the central nervous system, demeclocycline has become the preferred agent. Given in doses of 300 to 600 mg twice daily, demeclocycline promotes free water excretion, but often takes 1 to 2 weeks of therapy to begin working.
Renal failure due to demeclocycline has been reported in patients with concomitant liver disease.23 Demeclocycline can also cause photosensitivity and is contraindicated in children and pregnant women due to abnormalities in bone and enamel formation. In addition, it can be expensive and may not be covered fully by some prescription plans.
Vasopressin receptor antagonists (‘vaptans’)
ADH, also called vasopressin, interacts with various receptor subtypes, including V1a (causing vasoconstriction, platelet aggregation, inotropic stimulation, myocardial protein synthesis), V1b (causing secretion of adrenocorticotropic hormone), and V2 (causing water reabsorption and release of von Willebrand factor and factor VIII).
Conivaptan (Vaprisol) is a combined V1a-V2 antagonist that has been approved for the treatment of euvolemic and hypervolemic hyponatremia. Conivaptan inhibits the cytochrome P450 3A4 system and thus may interact with other drugs; therefore, its use has been limited to no more than 4 days of intravenous administration in the hospital setting. The recommended dosage is an initial 20-mg infusion over 30 minutes, followed by continuous infusions of 20 to 40 mg/day. Dosing adjustments in renal and hepatic impairment have not been well defined.
Tolvaptan (Samsca) is an oral selective V2 antagonist that has been studied in patients with euvolemic and hypervolemic hyponatremia. 26 Studies have included patients with congestive heart failure, cirrhosis, and SIADH. Although tolvaptan has not been shown to reduce rates of rehospitalization or death in congestive heart failure, it improves serum sodium, overall fluid balance, and congestive symptoms.27 Tolvaptan has recently been approved for the treatment of euvolemic and hypervolemic hyponatremia.
A recent study has confirmed the longterm efficacy of tolvaptan in 111 patients over a mean duration of treatment greater than 700 days.28 While the clinical benefits of chronic tolvaptan therapy have yet to be clearly demonstrated, this study shows that tolvaptan therapy can result in a sustained improvement in serum sodium concentration without an unacceptable increase in adverse events.29
Lixivaptan (VPA-985), another oral selective V2 receptor antagonist, is being studied in patients with euvolemic and hypervolemic hyponatremia.
Current role of vasopressin antagonists
Current studies of vasopressin antagonists in the treatment of hyponatremia are promising, though definite recommendations are needed to ensure slow, careful correction of hyponatremia. Most studies suggest that these agents provide slow, reliable increases in serum sodium. In one large study of patients with congestive heart failure, serum sodium rose by more than 12 mmol/L in 24 hours in fewer than 2% of patients.26
Notably, no cases of osmotic demyelination syndrome have been reported in these studies. However, it should be noted that therapy was started in the hospital with close monitoring of serum sodium levels and discontinuation of fluid restriction; the incidence of overly rapid correction of sodium may be higher outside of carefully done clinical studies. Clinicians should adopt monitoring strategies similar to those used in these rigorous studies.
At present, there is little experience with vasopressin antagonists in hyponatremic patients with serious signs or symptoms of cerebral edema, and most clinicians still view 3% saline as the gold standard for these patients.
Vasopressin antagonists should not be used in patients with hypovolemic hyponatremia, due to concerns about V1a blockade causing hypotension and about V2 blockade producing water excretion and a worsening of the volume-depleted state.
Recent clinical trials have reported that patients often experience increased thirst while taking these agents. This highlights the need to monitor serum sodium during treatment.
These agents are expensive. Tolvaptan costs about $250 per tablet; conivaptan, which is administered intravenously, may cost a little more per treatment course.
THERAPY IN SPECIFIC DISEASE STATES
Patients with hyponatremia and cirrhosis
The focus of treatment remains water and salt restriction and judicious use of loop diuretics and aldosterone antagonists such as spironolactone (Aldactone).
Tolvaptan has been effective at raising the serum sodium level in patients with cirrhosis, 26 while conivaptan should be avoided at present because of vasodilation from V1a receptor antagonism and its potential effects on systemic hemodynamics and risk of variceal bleeding.30
As the severity of cirrhosis increases, the only effective treatment of hyponatremia is liver transplantation.
Patients with SIADH
In most cases, water restriction is the mainstay of therapy. Adequate nutritional intake should also be stressed so that enough solute is available for ongoing water excretion. Although fluid restriction is usually effective, many patients cannot adhere to the level of restriction required.
In cases in which fluid restriction is not effective on its own, demeclocyline can be used to antagonize ADH action and increase water excretion. Sodium tablets and loop diuretics can also be used, taking care to avoid hypovolemia from diuretic-induced sodium losses. The use of tolvaptan in patients with SIADH has resulted in short-term increases in serum sodium.26 A recent study has suggested that this effect can be sustained with longer-term treatment,28 but further studies are needed to show a complementary clinical benefit (eg, improved neurocognition) to guide the use of these costly agents in clinical practice.
Patients with diuretic-induced hyponatremia
Thiazide diuretics should be discontinued and hypovolemia and hypokalemia should be corrected with isotonic saline and potassium supplementation. As the hypokalemia is corrected and the diuretic effect and hypovolemic stimulus to ADH dissipates, water excretion can increase rapidly, resulting in a brisk change in serum sodium.
Serum sodium levels should be closely monitored during therapy to avoid overcorrection. For this reason, use of hypertonic saline should generally be avoided. Hypotonic fluid— eg, half-normal (0.45%) or quarter-normal (0.22%) saline or even desmopressin—may become necessary in the later stages of therapy to avoid overly rapid correction.
Patients with exercise-associated hyponatremia
Patients at highest risk of exercise-associated hyponatremia include those who drink too much fluid during a long-distance race, who have low body weight, who are female, who exercise longer than 4 hours, and who use nonsteroidal anti-inflammatory drugs.31 The cause of hyponatremia is likely multifactorial, with excessive water intake coupled with sodium losses and impaired renal excretion of water due to ADH action and impaired renal dilution. To prevent exercise-associated hyponatremia, fluid intake should be limited to 400 to 800 mL/hour, with the higher end recommended for larger athletes and hotter climates.
Consensus recommendations suggest that most patients with mild hyponatremia (serum sodium 130 to 135 mmol/L) should be treated with fluid restriction and clinical observation, as spontaneous water diuresis leads to improvement in the serum sodium level. Importantly, the reflex to provide isotonic saline infusions should be avoided unless clear signs of volume depletion are present. Intravenous saline has the potential to worsen hyponatremia in the presence of ADH. In addition, some athletes will have retained water in the gastrointestinal tract that may be mobilized after the race, resulting in worsening of hyponatremia.32
In athletes with severe hyponatremia (serum sodium < 120 mmol/L) or symptomatic exercise-associated hyponatremia (lethargy, respiratory depression, seizures), hypertonic saline is the treatment of choice. One protocol suggests giving 100 mL of 3% saline over 10 minutes in the field, followed by prompt transportation to hospital.33
SUMMARY POINTS
- Hyponatremia is a common electrolyte disorder that in its most severe form requires urgent therapy with hypertonic saline to correct cerebral edema.
- In patients without serious signs or symptoms of cerebral edema, recent observations suggest there may be clinically important symptomatology relating to mild neurocognitive dysfunction and an association with risk of bone fracture.
- Multiple treatment strategies are available according to the underlying extracellular fluid volume status and cause of hyponatremia. These include fluid and sodium restriction and augmentation of urinary water excretion with various nutritional and pharmacologic strategies. The most novel therapy includes antagonism of the vasopressin V2 receptor with a class of aquaretic agents known as vaptans.
- There can be serious neurologic injury associated with overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia.
- Clinicians must be familiar with the details of each of the treatments and have an appreciation of the importance of careful monitoring during treatment.
- Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet 1981; 2:26–31.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med 2007; 120(suppl 1):S1–S21.
- Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med 2004; 71:639–650.
- Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med 1989; 86:315–318.
- Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant 2003; 18:2486–2491.
- Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med 2007; 74:377–383.
- Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol 1987; 252:F661–F669.
- Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol 1997; 8:1599–1607.
- Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int 2006; 69:1291–1293.
- Ellis SJ. Severe hyponatraemia: complications and treatment. QJM 1995; 88:905–909.
- Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol 1994; 4:1522–1530.
- Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol 2008; 3:331–336.
- Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int 2009; 76:587–589.
- Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol 2004; 8:12–16.
- Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med 2000; 342:1581–1589.
- Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol 2007; 2:1110–1117.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med 2007; 356:2064–2072.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med 2006; 119:71.e1–e8.
- Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol 2010; 5:275–280.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med 1987; 83:905–988.
- Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM 2005; 98:529–540.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol 2008; 19:1076–1078.
- Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med 1977; 87:195–197.
- Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists? J Am Soc Nephrol 2008; 19:1054–1058.
- Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet 2008; 371:1624–1632.
- Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 2006; 355:2099–2112.
- Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007; 297:1319–1331.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol 2010; 21:705–712.
- Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if? J Am Soc Nephrol 2010; 21:552–555.
- Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int 2006; 69:2124–2130.
- Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol 2007; 2:151–161.
- Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med 2005; 353:427–428.
- Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med 2005; 15:208–213.
- Flear CTG, Gill GV, Burn J. Hyponatremia: mechanisms and management. Lancet 1981; 2:26–31.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med 2007; 120(suppl 1):S1–S21.
- Freda BJ, Davidson MB, Hall PM. Evaluation of hyponatremia: a little physiology goes a long way. Cleve Clin J Med 2004; 71:639–650.
- Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med 1989; 86:315–318.
- Moritz L, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant 2003; 18:2486–2491.
- Widdess-Walsh P, Sabharwal V, Demirjian S, DeGeorgia M. Neurologic effects of hyponatremia and its treatment. Cleve Clin J Med 2007; 74:377–383.
- Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol 1987; 252:F661–F669.
- Lauriat SM, Berl T. The hyponatremic patient: practical focus on therapy. J Am Soc Nephrol 1997; 8:1599–1607.
- Kokko JP. Symptomatic hyponatremia with hypoxia is a medical emergency. Kidney Int 2006; 69:1291–1293.
- Ellis SJ. Severe hyponatraemia: complications and treatment. QJM 1995; 88:905–909.
- Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol 1994; 4:1522–1530.
- Perianayagam A, Sterns RH, Silver SM, et al. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol 2008; 3:331–336.
- Sterns RH, Hix JK. Overcorrection of hyponatremia is a medical emergency. Kidney Int 2009; 76:587–589.
- Nguyen MK, Kurtz I. Analysis of current formulas used for treatment of the dysnatremias. Clin Exp Nephrol 2004; 8:12–16.
- Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med 2000; 342:1581–1589.
- Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol 2007; 2:1110–1117.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med 2007; 356:2064–2072.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med 2006; 119:71.e1–e8.
- Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol 2010; 5:275–280.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med 1987; 83:905–988.
- Hoorn EJ, Halperin ML, Zietse R. Diagnostic approach to a patient with hyponatraemia: traditional versus physiology-based options. QJM 2005; 98:529–540.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol 2008; 19:1076–1078.
- Carrilho F, Bosch J, Arroyo V, Mas A, Viver J, Rodes J. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med 1977; 87:195–197.
- Lehrich RW, Greenberg A. When is it appropriate to use vasopressin receptor antagonists? J Am Soc Nephrol 2008; 19:1054–1058.
- Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet 2008; 371:1624–1632.
- Schrier RW, Gross P, Gheorghiade M, et al; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 2006; 355:2099–2112.
- Konstam MA, Gheorghiade M, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007; 297:1319–1331.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol 2010; 21:705–712.
- Greenberg A, Lehrich RW. Treatment of chronic hyponatremia: now we know how, but do we know when or if? J Am Soc Nephrol 2010; 21:552–555.
- Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int 2006; 69:2124–2130.
- Rosner MH, Kirven J. Exercise-associated hyponatremia. Clin J Am Soc Nephrol 2007; 2:151–161.
- Halperin ML, Kamel KS, Sterns R. Hyponatremia in marathon runners. N Engl J Med 2005; 353:427–428.
- Hew-Butler T, Almond C, Ayus JC, et al; Exercise-Associated Hyponatremia (EAH) Consensus Panel. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med 2005; 15:208–213.
KEY POINTS
- Some hyponatremic patients present with acute, life-threatening cerebral edema due to severe hyponatremia. In others, the hyponatremia may be chronic and less severe, causing relatively few symptoms, but representing an important, independent marker of poor prognosis due to an underlying disease (eg, heart failure).
- Even patients with chronic, less severe hyponatremia may have subtle symptoms of neurocognitive dysfunction and a higher risk of bone fractures.
- Overly rapid correction of chronic hyponatremia or undercorrection of acute symptomatic hyponatremia can lead to serious neurologic injury.
- Treatment strategies vary depending on the extracellular fluid volume status and the cause of hyponatremia.
- Vasopressin antagonists (“vaptans”), a new class of aquaretic agents, specifically target the mechanism driving hyponatremia in some patients.