User login
Prazosin for PTSD: Sorting out the evidence

Mr. H, age 43, presents to your clinic for management of posttraumatic stress disorder (PTSD). At his last appointment 8 weeks ago, he was continued on fluoxetine, 60 mg/d; he had been stable on this medication for 6 months. Today, Mr. H reports an increase in the frequency and severity of nightmares. He states that he wakes at least 3 times every week with “disturbing dreams” about his time in the military and does not feel rested even when he sleeps through the night. His Clinician-Administered PTSD Scale (CAPS) score is 95 on this visit, suggesting extreme PTSD symptomatology. Mr. H asks if anything can be done to reduce the frequency and intensity of his nightmares.
PTSD is the development of characteristic symptoms following exposure to ≥1 traumatic events. According to DSM-5, PTSD symptoms include the presence of ≥1 intrusion symptoms (recurrent, intrusive memories of the traumatic event; recurrent distressing dreams; dissociative reactions), persistent avoidance of stimuli, negative alterations in cognition and mood, and marked alterations in arousal and reactivity associated with the traumatic event(s).1 The symptoms must be present for >1 month, cause clinically significant distress or impairment in functioning, and not be attributable to the psychologic effects of a substance or medical conditions.1 This article focuses specifically on the hyperarousal symptoms, and the clinical controversies surrounding the use of prazosin for PTSD.
Prazosin for PTSD treatment
Sleep disorders are extremely common in patients with PTSD. Up to 90% of patients report sleep disturbances, and up to 70% report nightmares.2 Prazosin has been widely used in the treatment of PTSD-related sleep disorders and nightmares.The American Psychiatric Association3 and the British Association of Psychopharmacology4 guidelines in-clude prazosin as a first-line recommendation for treatment of PTSD. However, updated 2017 guidelines from the Veterans Affairs/Department of Defense (VA/DoD)5 and data from the 2018 Prazosin and Combat Trauma PTSD (PACT) trial6 contradict these original recommendations. Previously, the 2010 VA/DoD guideline said prazosin had insufficient evidence for monotherapy, but recommended it as adjunctive treatment for sleep and nightmares.7 The updated 2017 VA/DoD guideline recommends “weak against” prazosin use for global symptoms of PTSD, and says there is insufficient evidence for its use in nightmares.5 Below we summarize the findings of studies that contributed to those original recommendations, along with results of the PACT trial.
Raskind et al8,9 conducted 2 studies of prazosin use in combat veterans with PTSD. In both studies, prazosin had significant positive effects on the Clinician-Administered PTSD Scale (CAPS) and Clinical Global Impression of Change (CGIC) scores.8,9 The 2007 study also found significant effects of prazosin on Pittsburgh Sleep Quality Index (PSQI) scores.9
Raskind et al10 conducted another study in 2013 of prazosin use for active-duty soldiers who had combat trauma PTSD with nightmares. Prazosin had positive effects for nightmares, sleep quality, and CAPS scores.10
Germain et al11 reviewed prazosin for treating sleep disturbances in US military veterans. Prazosin was associated with significant improvements in insomnia and daytime PTSD symptom severity as demonstrated by changes in PSQI and CAPS scores.11
Taylor et al12 examined the effects of prazosin on sleep measures and clinical symptoms in civilians with PTSD. Prazosin significantly increased total sleep time, rapid eye movement sleep time, and CGIC scores while significantly decreasing trauma-related nightmares.12
Continue to: Overall, these trials...
Overall, these trials found efficacy for the use of prazosin for patients diagnosed with PTSD; however, the population size in each of these studies was small.
Results of the PACT trial
The PACT trial was a 26-week, multicenter, double-blind, randomized, placebo-controlled trial conducted across 12 VA medical centers.6 During the first 5 weeks, participants were randomized to receive placebo or prazosin, which could be titrated up to 20 mg/d in men and 12 mg/d in women. Participants remained on that dose from the end of Week 5 through Week 10. At that time, other pharmacologic therapies and psychotherapy could be added, discontinued, or adjusted. The mean maintenance total daily dose of prazosin was 14.8 mg.
A total of 413 patients were screened, 304 were randomized (152 per group), and 271 completed the 10-week primary outcome assessment. The population was almost entirely male (96.1% in the prazosin group and 99.3% in the placebo group), and most participants were White (64.5% in the prazosin group and 69.1% in the placebo group), with an average age of approximately 50 years. Primary outcomes included change from baseline to Week 10 in both CAPS item B2 (“recurrent distressing dreams”) and PSQI scores. CGIC score was evaluated at Week 10.
At Week 10, none of the primary outcomes were found to be statistically significant. The mean difference in change from baseline to Week 10 in CAPS item B2 score and PSQI score were 0.2 (P = .38) and 0.1 (P = .80), respectively. There was no significant difference in mean CGIC scores (P = .96). Repeated measures of CAPS item B2, PSQI, and CGIC scores were conducted through Week 26 as secondary outcomes. No significant differences were found. This study concluded that prazosin did not alleviate distressing dreams, improve sleep quality, or improve overall clinical symptoms.6
The PACT trial: Strengths and weaknesses
The PACT trial is the largest placebo-controlled trial for prazosin use in PTSD to date. It failed to show efficacy of prazosin for PTSD-associated nightmares, which contradicts previous studies. Although the mean total daily dose of prazosin was adequate and primary outcomes were measured with appropriate scales, the study failed to enroll the desired number of patients, which increased the possibility of false-negative results. Furthermore, participant recruitment may have led to selection bias because all participants were clinically stable, which could explain the lack of efficacy. However, the average CAPS scores were 80.7 in the prazosin group and 81.9 in the placebo group, which indicates that these patients had significant symptomatology at baseline and before entering the study.
Continue to: A major theme...
A major theme of studies evaluating prazosin treatment for PTSD is a focus on a military population and military-related trauma. Other than Taylor et al12 (N=13), none of these trials included patients who were diagnosed with PTSD due to other traumas, such as sexual trauma, which limits the generalizability of the results. Furthermore, apart from the PACT trial, none of these studies had >100 participants, which further reduces external validity. Current guidelines have not been updated to include the results of the PACT trial, and it is unclear if the results of this trial are strong enough to change clinical practice.
CASE CONTINUED
To ensure patient-centered care, the treating clinicians conduct a risk/benefit discussion with the patient regarding starting prazosin. Mr. H opts to try prazosin, so the clinicians initiate a low dose (1 mg/d) to mitigate adverse effects, and plan to titrate to clinical effect or intolerability. Per evidence from the trials discussed, it is likely Mr. H will need to be titrated to at least 5 to 6 mg/d to see a clinical effect.
Related Resource
North CS, Hong BA, Downs DL. PTSD: A systematic approach to diagnosis and treatment. Current Psychiatry 2018;17(4):35-43.
Drug Brand Names
Fluoxetine • Prozac
Prazosin • Minipress
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Maher MJ, Rego SA, Asnis, GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
3. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. APA Practice Guidelines. Published 2010. Accessed March 14, 2021. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/acutestressdisorderptsd-watch.pdf
4. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439. doi: 10.1177/0269881114525674
5. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0. Published 2017. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal012418.pdf
6. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
7. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline: management of post-traumatic stress. Version 2.0. Published 2010. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/cpg_PTSD-full-201011612.PDF
8. Raskind MA, Peskind ER, Katner ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo-controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
10. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
11. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
12. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63(6):629-632.
Mr. H, age 43, presents to your clinic for management of posttraumatic stress disorder (PTSD). At his last appointment 8 weeks ago, he was continued on fluoxetine, 60 mg/d; he had been stable on this medication for 6 months. Today, Mr. H reports an increase in the frequency and severity of nightmares. He states that he wakes at least 3 times every week with “disturbing dreams” about his time in the military and does not feel rested even when he sleeps through the night. His Clinician-Administered PTSD Scale (CAPS) score is 95 on this visit, suggesting extreme PTSD symptomatology. Mr. H asks if anything can be done to reduce the frequency and intensity of his nightmares.
PTSD is the development of characteristic symptoms following exposure to ≥1 traumatic events. According to DSM-5, PTSD symptoms include the presence of ≥1 intrusion symptoms (recurrent, intrusive memories of the traumatic event; recurrent distressing dreams; dissociative reactions), persistent avoidance of stimuli, negative alterations in cognition and mood, and marked alterations in arousal and reactivity associated with the traumatic event(s).1 The symptoms must be present for >1 month, cause clinically significant distress or impairment in functioning, and not be attributable to the psychologic effects of a substance or medical conditions.1 This article focuses specifically on the hyperarousal symptoms, and the clinical controversies surrounding the use of prazosin for PTSD.
Prazosin for PTSD treatment
Sleep disorders are extremely common in patients with PTSD. Up to 90% of patients report sleep disturbances, and up to 70% report nightmares.2 Prazosin has been widely used in the treatment of PTSD-related sleep disorders and nightmares.The American Psychiatric Association3 and the British Association of Psychopharmacology4 guidelines in-clude prazosin as a first-line recommendation for treatment of PTSD. However, updated 2017 guidelines from the Veterans Affairs/Department of Defense (VA/DoD)5 and data from the 2018 Prazosin and Combat Trauma PTSD (PACT) trial6 contradict these original recommendations. Previously, the 2010 VA/DoD guideline said prazosin had insufficient evidence for monotherapy, but recommended it as adjunctive treatment for sleep and nightmares.7 The updated 2017 VA/DoD guideline recommends “weak against” prazosin use for global symptoms of PTSD, and says there is insufficient evidence for its use in nightmares.5 Below we summarize the findings of studies that contributed to those original recommendations, along with results of the PACT trial.
Raskind et al8,9 conducted 2 studies of prazosin use in combat veterans with PTSD. In both studies, prazosin had significant positive effects on the Clinician-Administered PTSD Scale (CAPS) and Clinical Global Impression of Change (CGIC) scores.8,9 The 2007 study also found significant effects of prazosin on Pittsburgh Sleep Quality Index (PSQI) scores.9
Raskind et al10 conducted another study in 2013 of prazosin use for active-duty soldiers who had combat trauma PTSD with nightmares. Prazosin had positive effects for nightmares, sleep quality, and CAPS scores.10
Germain et al11 reviewed prazosin for treating sleep disturbances in US military veterans. Prazosin was associated with significant improvements in insomnia and daytime PTSD symptom severity as demonstrated by changes in PSQI and CAPS scores.11
Taylor et al12 examined the effects of prazosin on sleep measures and clinical symptoms in civilians with PTSD. Prazosin significantly increased total sleep time, rapid eye movement sleep time, and CGIC scores while significantly decreasing trauma-related nightmares.12
Continue to: Overall, these trials...
Overall, these trials found efficacy for the use of prazosin for patients diagnosed with PTSD; however, the population size in each of these studies was small.
Results of the PACT trial
The PACT trial was a 26-week, multicenter, double-blind, randomized, placebo-controlled trial conducted across 12 VA medical centers.6 During the first 5 weeks, participants were randomized to receive placebo or prazosin, which could be titrated up to 20 mg/d in men and 12 mg/d in women. Participants remained on that dose from the end of Week 5 through Week 10. At that time, other pharmacologic therapies and psychotherapy could be added, discontinued, or adjusted. The mean maintenance total daily dose of prazosin was 14.8 mg.
A total of 413 patients were screened, 304 were randomized (152 per group), and 271 completed the 10-week primary outcome assessment. The population was almost entirely male (96.1% in the prazosin group and 99.3% in the placebo group), and most participants were White (64.5% in the prazosin group and 69.1% in the placebo group), with an average age of approximately 50 years. Primary outcomes included change from baseline to Week 10 in both CAPS item B2 (“recurrent distressing dreams”) and PSQI scores. CGIC score was evaluated at Week 10.
At Week 10, none of the primary outcomes were found to be statistically significant. The mean difference in change from baseline to Week 10 in CAPS item B2 score and PSQI score were 0.2 (P = .38) and 0.1 (P = .80), respectively. There was no significant difference in mean CGIC scores (P = .96). Repeated measures of CAPS item B2, PSQI, and CGIC scores were conducted through Week 26 as secondary outcomes. No significant differences were found. This study concluded that prazosin did not alleviate distressing dreams, improve sleep quality, or improve overall clinical symptoms.6
The PACT trial: Strengths and weaknesses
The PACT trial is the largest placebo-controlled trial for prazosin use in PTSD to date. It failed to show efficacy of prazosin for PTSD-associated nightmares, which contradicts previous studies. Although the mean total daily dose of prazosin was adequate and primary outcomes were measured with appropriate scales, the study failed to enroll the desired number of patients, which increased the possibility of false-negative results. Furthermore, participant recruitment may have led to selection bias because all participants were clinically stable, which could explain the lack of efficacy. However, the average CAPS scores were 80.7 in the prazosin group and 81.9 in the placebo group, which indicates that these patients had significant symptomatology at baseline and before entering the study.
Continue to: A major theme...
A major theme of studies evaluating prazosin treatment for PTSD is a focus on a military population and military-related trauma. Other than Taylor et al12 (N=13), none of these trials included patients who were diagnosed with PTSD due to other traumas, such as sexual trauma, which limits the generalizability of the results. Furthermore, apart from the PACT trial, none of these studies had >100 participants, which further reduces external validity. Current guidelines have not been updated to include the results of the PACT trial, and it is unclear if the results of this trial are strong enough to change clinical practice.
CASE CONTINUED
To ensure patient-centered care, the treating clinicians conduct a risk/benefit discussion with the patient regarding starting prazosin. Mr. H opts to try prazosin, so the clinicians initiate a low dose (1 mg/d) to mitigate adverse effects, and plan to titrate to clinical effect or intolerability. Per evidence from the trials discussed, it is likely Mr. H will need to be titrated to at least 5 to 6 mg/d to see a clinical effect.
Related Resource
North CS, Hong BA, Downs DL. PTSD: A systematic approach to diagnosis and treatment. Current Psychiatry 2018;17(4):35-43.
Drug Brand Names
Fluoxetine • Prozac
Prazosin • Minipress
Mr. H, age 43, presents to your clinic for management of posttraumatic stress disorder (PTSD). At his last appointment 8 weeks ago, he was continued on fluoxetine, 60 mg/d; he had been stable on this medication for 6 months. Today, Mr. H reports an increase in the frequency and severity of nightmares. He states that he wakes at least 3 times every week with “disturbing dreams” about his time in the military and does not feel rested even when he sleeps through the night. His Clinician-Administered PTSD Scale (CAPS) score is 95 on this visit, suggesting extreme PTSD symptomatology. Mr. H asks if anything can be done to reduce the frequency and intensity of his nightmares.
PTSD is the development of characteristic symptoms following exposure to ≥1 traumatic events. According to DSM-5, PTSD symptoms include the presence of ≥1 intrusion symptoms (recurrent, intrusive memories of the traumatic event; recurrent distressing dreams; dissociative reactions), persistent avoidance of stimuli, negative alterations in cognition and mood, and marked alterations in arousal and reactivity associated with the traumatic event(s).1 The symptoms must be present for >1 month, cause clinically significant distress or impairment in functioning, and not be attributable to the psychologic effects of a substance or medical conditions.1 This article focuses specifically on the hyperarousal symptoms, and the clinical controversies surrounding the use of prazosin for PTSD.
Prazosin for PTSD treatment
Sleep disorders are extremely common in patients with PTSD. Up to 90% of patients report sleep disturbances, and up to 70% report nightmares.2 Prazosin has been widely used in the treatment of PTSD-related sleep disorders and nightmares.The American Psychiatric Association3 and the British Association of Psychopharmacology4 guidelines in-clude prazosin as a first-line recommendation for treatment of PTSD. However, updated 2017 guidelines from the Veterans Affairs/Department of Defense (VA/DoD)5 and data from the 2018 Prazosin and Combat Trauma PTSD (PACT) trial6 contradict these original recommendations. Previously, the 2010 VA/DoD guideline said prazosin had insufficient evidence for monotherapy, but recommended it as adjunctive treatment for sleep and nightmares.7 The updated 2017 VA/DoD guideline recommends “weak against” prazosin use for global symptoms of PTSD, and says there is insufficient evidence for its use in nightmares.5 Below we summarize the findings of studies that contributed to those original recommendations, along with results of the PACT trial.
Raskind et al8,9 conducted 2 studies of prazosin use in combat veterans with PTSD. In both studies, prazosin had significant positive effects on the Clinician-Administered PTSD Scale (CAPS) and Clinical Global Impression of Change (CGIC) scores.8,9 The 2007 study also found significant effects of prazosin on Pittsburgh Sleep Quality Index (PSQI) scores.9
Raskind et al10 conducted another study in 2013 of prazosin use for active-duty soldiers who had combat trauma PTSD with nightmares. Prazosin had positive effects for nightmares, sleep quality, and CAPS scores.10
Germain et al11 reviewed prazosin for treating sleep disturbances in US military veterans. Prazosin was associated with significant improvements in insomnia and daytime PTSD symptom severity as demonstrated by changes in PSQI and CAPS scores.11
Taylor et al12 examined the effects of prazosin on sleep measures and clinical symptoms in civilians with PTSD. Prazosin significantly increased total sleep time, rapid eye movement sleep time, and CGIC scores while significantly decreasing trauma-related nightmares.12
Continue to: Overall, these trials...
Overall, these trials found efficacy for the use of prazosin for patients diagnosed with PTSD; however, the population size in each of these studies was small.
Results of the PACT trial
The PACT trial was a 26-week, multicenter, double-blind, randomized, placebo-controlled trial conducted across 12 VA medical centers.6 During the first 5 weeks, participants were randomized to receive placebo or prazosin, which could be titrated up to 20 mg/d in men and 12 mg/d in women. Participants remained on that dose from the end of Week 5 through Week 10. At that time, other pharmacologic therapies and psychotherapy could be added, discontinued, or adjusted. The mean maintenance total daily dose of prazosin was 14.8 mg.
A total of 413 patients were screened, 304 were randomized (152 per group), and 271 completed the 10-week primary outcome assessment. The population was almost entirely male (96.1% in the prazosin group and 99.3% in the placebo group), and most participants were White (64.5% in the prazosin group and 69.1% in the placebo group), with an average age of approximately 50 years. Primary outcomes included change from baseline to Week 10 in both CAPS item B2 (“recurrent distressing dreams”) and PSQI scores. CGIC score was evaluated at Week 10.
At Week 10, none of the primary outcomes were found to be statistically significant. The mean difference in change from baseline to Week 10 in CAPS item B2 score and PSQI score were 0.2 (P = .38) and 0.1 (P = .80), respectively. There was no significant difference in mean CGIC scores (P = .96). Repeated measures of CAPS item B2, PSQI, and CGIC scores were conducted through Week 26 as secondary outcomes. No significant differences were found. This study concluded that prazosin did not alleviate distressing dreams, improve sleep quality, or improve overall clinical symptoms.6
The PACT trial: Strengths and weaknesses
The PACT trial is the largest placebo-controlled trial for prazosin use in PTSD to date. It failed to show efficacy of prazosin for PTSD-associated nightmares, which contradicts previous studies. Although the mean total daily dose of prazosin was adequate and primary outcomes were measured with appropriate scales, the study failed to enroll the desired number of patients, which increased the possibility of false-negative results. Furthermore, participant recruitment may have led to selection bias because all participants were clinically stable, which could explain the lack of efficacy. However, the average CAPS scores were 80.7 in the prazosin group and 81.9 in the placebo group, which indicates that these patients had significant symptomatology at baseline and before entering the study.
Continue to: A major theme...
A major theme of studies evaluating prazosin treatment for PTSD is a focus on a military population and military-related trauma. Other than Taylor et al12 (N=13), none of these trials included patients who were diagnosed with PTSD due to other traumas, such as sexual trauma, which limits the generalizability of the results. Furthermore, apart from the PACT trial, none of these studies had >100 participants, which further reduces external validity. Current guidelines have not been updated to include the results of the PACT trial, and it is unclear if the results of this trial are strong enough to change clinical practice.
CASE CONTINUED
To ensure patient-centered care, the treating clinicians conduct a risk/benefit discussion with the patient regarding starting prazosin. Mr. H opts to try prazosin, so the clinicians initiate a low dose (1 mg/d) to mitigate adverse effects, and plan to titrate to clinical effect or intolerability. Per evidence from the trials discussed, it is likely Mr. H will need to be titrated to at least 5 to 6 mg/d to see a clinical effect.
Related Resource
North CS, Hong BA, Downs DL. PTSD: A systematic approach to diagnosis and treatment. Current Psychiatry 2018;17(4):35-43.
Drug Brand Names
Fluoxetine • Prozac
Prazosin • Minipress
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Maher MJ, Rego SA, Asnis, GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
3. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. APA Practice Guidelines. Published 2010. Accessed March 14, 2021. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/acutestressdisorderptsd-watch.pdf
4. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439. doi: 10.1177/0269881114525674
5. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0. Published 2017. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal012418.pdf
6. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
7. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline: management of post-traumatic stress. Version 2.0. Published 2010. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/cpg_PTSD-full-201011612.PDF
8. Raskind MA, Peskind ER, Katner ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo-controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
10. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
11. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
12. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63(6):629-632.
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Maher MJ, Rego SA, Asnis, GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
3. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. APA Practice Guidelines. Published 2010. Accessed March 14, 2021. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/acutestressdisorderptsd-watch.pdf
4. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439. doi: 10.1177/0269881114525674
5. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0. Published 2017. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal012418.pdf
6. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
7. Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline: management of post-traumatic stress. Version 2.0. Published 2010. Accessed February 5, 2021. https://www.healthquality.va.gov/guidelines/MH/ptsd/cpg_PTSD-full-201011612.PDF
8. Raskind MA, Peskind ER, Katner ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo-controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
10. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
11. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
12. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63(6):629-632.

Management of major depressive disorder with psychotic features

Mrs. C, age 56, has a history of major depressive disorder (MDD). She has been stable for 5 years without medication. Six months ago, she presented to you, along with her son, seeking help. She reported that she had been experiencing insomnia, fatigue, and was not engaging in hobbies. Her son told you that his mother had lost weight and had been avoiding family dinners. Mrs. C reported recurrent thoughts of dying and heard voices vividly telling her that she was a burden and that her family would be better off without her. However, there was no imminent danger of self-harm. At that appointment, you initiated
Since that time, Mrs. C has followed up with you monthly with good response to the medications. Currently, she states her depression is much improved, and she denies hearing voices for approximately 5 months.
Based on her presentation and response, what do the data suggest about her length of treatment, and when should you consider tapering the antipsychotic medication?
In DSM-5, MDD with psychotic features is a severe subtype of MDD that is defined as a major depressive episode characterized by delusions and/or hallucinations.1 In the general population, the lifetime prevalence of this disorder varies from 0.35% to 1%, and the rate is higher in older patients.2 Risk factors include female gender, family history, and concomitant bipolar disorder.2
Epidemiologic studies have shown that psychotic features can occur in 15% to 20% of patients with MDD. The psychotic features that occur during these episodes are delusions and hallucinations.1 These features can be either mood-congruent (related to the depressive themes of worthlessness or guilt) or mood-incongruent (ie, unrelated to depressive themes).1
Treatment options: ECT or pharmacotherapy
Guidelines from the American Psychiatric Association3 and the National Institute for Clinical Excellence4 recommend treating depression with psychosis with electroconvulsive therapy (ECT) or with combined antidepressant and antipsychotic medications as first-line options. The Texas Medication Algorithm Project (TMAP) Algorithm for MDD,5 which closely focuses on treatment of MDD with psychotic features, can be used for treatment decisions (see Related Resources).
Electroconvulsive therapy is known to be efficacious in treating patients with MDD with psychotic features and should be considered as a treatment option. However, medication therapy is often chosen as the initial treatment due to the limitations of ECT, including accessibility, cost, and patient preference. However, in certain cases, ECT is the preferred option because it can provide rapid and significant improvement in patients with severe psychosis, suicidality, or risk of imminent harm.
Continue to: Pharmacotherapy
Pharmacotherapy for the treatment of MDD with psychotic features should consist of a combination of an antidepressant and antipsychotic medication. This combination has been shown to be more effective than either agent alone. Some combinations have been studied specifically for MDD with psychosis. The Study of the Pharmacotherapy of Psychotic Depression (STOP-PD), a 12-week, double-blind, randomized controlled trial, found that the combination of sertraline and olanzapine was efficacious and superior to monotherapy with olanzapine in an acute setting.6 In another study, the combination of olanzapine and
How long should treatment last?
The optimal timeline for treating patients with MDD with psychotic features is unknown. According to the TMAP algorithm and expert opinion, the continuation phase of pharmacotherapy should include treatment for at least 4 months with an antipsychotic medication and at least 2 years to lifetime treatment with an antidepressant.5 The STOP-PD II study, which was a continuation of the 12-week STOP-PD study, examined antipsychotic duration to determine the effects of continuing olanzapine once an episode of psychotic depression had responded to olanzapine and sertraline.11 Patients who had achieved remission after receiving olanzapine and sertraline were randomized to continue to receive this combination or to receive sertraline plus placebo for 36 weeks. The primary outcome was relapse, which was broadly defined as 1 of the following11:
- a Structured Clinical Interview for the DSM (SCID)-rated assessment that revealed the patient had enough symptoms to meet criteria for a DSM-IV major depressive episode
- a 17-item HAM-D scoren of ≥18
- SCID-rated psychosis
- other significant clinical worsening, defined as having a suicide plan or attempting suicide, developing SCID-rated symptoms of mania or hypomania, or being hospitalized in a psychiatric unit.
Compared with sertraline plus placebo, continuing sertraline plus olanzapine reduced the risk of relapse over 36 weeks (hazard ratio, 0.25; 95% confidence interval, 0.13 to 0.48; P < .001).11 However, as expected, the incidence of adverse effects such as weight gain and parkinsonism was higher in the olanzapine group. Therefore, it is important to consider the potential long-term adverse effects of continuing antipsychotic medications.
Weighing the evidence
Electroconvulsive therapy is considered a first-line treatment option for MDD with psychotic features; however, because of limitations associated with this approach, antidepressants plus antipsychotics are often utilized as an initial treatment.
Continue to: CASE
CASE CONTINUED
Based on the results of the STOP-PD and STOP-PD II trials, Mrs. C should be continued on sertraline plus olanzapine for at least another 3 to 6 months before an olanzapine taper should be considered. At that time, the risks and benefits of a taper vs continuing therapy should be considered. Given her history of MDD and the severity of this most recent episode, sertraline therapy should be continued for at least 2 years, and possibly indefinitely.
Related Resources
- Texas Medication Algorithm Project. Algorithm for the treatment of major depressive disorder with psychotic features. https://chsciowa.org/sites/chsciowa.org/files/resource/files/9_-_depression_med_algorithm_supplement.pdf
- Dold M, Bartova L, Kautzky A, et al. Psychotic features in patients with major depressive disorder: a report from the European Group for the Study of Resistant Depression. J Clin Psychiatry. 2019;80(1):17m12090. doi: 10.4088/ JCP.17m12090
- Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7): 622-631.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Jääskeläinen E, Juola T, Korpela H, et al. Epidemiology of psychotic depression - systematic review and meta-analysis. Psychol Med. 2018;48(6):905-918.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(4)(suppl):1-45.
4. National Institute for Clinical Excellence. Depression in adults: recognition and management: clinical guideline [CG90]. National Institute for Health and Clinical Excellence. Published October 28, 2009. Accessed January 12, 2021. https://www.nice.org.uk/guidance/cg90
5. Crimson ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry. 1999;60(3):142-156.
6. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy for psychotic depression -- the STOP-PD study. Arch Gen Psychiatry. 2009;66(8):838-847.
7. Rothschild AJ, Williamson DJ, Tohen MF, et al. A double-blind, randomized study of olanzapine and olanzapine/fluoxetine combination for major depression with psychotic features. J Clin Psychopharmacol. 2004;24(4):365-373.
8. Wijkstra J, Burger H, van den Broek WW, et al. Treatment of unipolar psychotic depression: a randomized, doubleblind study comparing imipramine, venlafaxine, and venlafaxine plus quetiapine. Acta Psychiatr Scand. 2010;21(3):190-200.
9. Muller-Siecheneder F, Muller M, Hillert A, et al. Risperidone versus haloperidol and amitriptyline in the treatment of patients with a combined psychotic and depressive syndrome. J Clin Psychopharm. 1998;18(2):111-120.
10. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacological treatment of delusional depression. Am J Psychiatry. 1985;142(4):430-436.
11. Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7):622-631.
Mrs. C, age 56, has a history of major depressive disorder (MDD). She has been stable for 5 years without medication. Six months ago, she presented to you, along with her son, seeking help. She reported that she had been experiencing insomnia, fatigue, and was not engaging in hobbies. Her son told you that his mother had lost weight and had been avoiding family dinners. Mrs. C reported recurrent thoughts of dying and heard voices vividly telling her that she was a burden and that her family would be better off without her. However, there was no imminent danger of self-harm. At that appointment, you initiated
Since that time, Mrs. C has followed up with you monthly with good response to the medications. Currently, she states her depression is much improved, and she denies hearing voices for approximately 5 months.
Based on her presentation and response, what do the data suggest about her length of treatment, and when should you consider tapering the antipsychotic medication?
In DSM-5, MDD with psychotic features is a severe subtype of MDD that is defined as a major depressive episode characterized by delusions and/or hallucinations.1 In the general population, the lifetime prevalence of this disorder varies from 0.35% to 1%, and the rate is higher in older patients.2 Risk factors include female gender, family history, and concomitant bipolar disorder.2
Epidemiologic studies have shown that psychotic features can occur in 15% to 20% of patients with MDD. The psychotic features that occur during these episodes are delusions and hallucinations.1 These features can be either mood-congruent (related to the depressive themes of worthlessness or guilt) or mood-incongruent (ie, unrelated to depressive themes).1
Treatment options: ECT or pharmacotherapy
Guidelines from the American Psychiatric Association3 and the National Institute for Clinical Excellence4 recommend treating depression with psychosis with electroconvulsive therapy (ECT) or with combined antidepressant and antipsychotic medications as first-line options. The Texas Medication Algorithm Project (TMAP) Algorithm for MDD,5 which closely focuses on treatment of MDD with psychotic features, can be used for treatment decisions (see Related Resources).
Electroconvulsive therapy is known to be efficacious in treating patients with MDD with psychotic features and should be considered as a treatment option. However, medication therapy is often chosen as the initial treatment due to the limitations of ECT, including accessibility, cost, and patient preference. However, in certain cases, ECT is the preferred option because it can provide rapid and significant improvement in patients with severe psychosis, suicidality, or risk of imminent harm.
Continue to: Pharmacotherapy
Pharmacotherapy for the treatment of MDD with psychotic features should consist of a combination of an antidepressant and antipsychotic medication. This combination has been shown to be more effective than either agent alone. Some combinations have been studied specifically for MDD with psychosis. The Study of the Pharmacotherapy of Psychotic Depression (STOP-PD), a 12-week, double-blind, randomized controlled trial, found that the combination of sertraline and olanzapine was efficacious and superior to monotherapy with olanzapine in an acute setting.6 In another study, the combination of olanzapine and
How long should treatment last?
The optimal timeline for treating patients with MDD with psychotic features is unknown. According to the TMAP algorithm and expert opinion, the continuation phase of pharmacotherapy should include treatment for at least 4 months with an antipsychotic medication and at least 2 years to lifetime treatment with an antidepressant.5 The STOP-PD II study, which was a continuation of the 12-week STOP-PD study, examined antipsychotic duration to determine the effects of continuing olanzapine once an episode of psychotic depression had responded to olanzapine and sertraline.11 Patients who had achieved remission after receiving olanzapine and sertraline were randomized to continue to receive this combination or to receive sertraline plus placebo for 36 weeks. The primary outcome was relapse, which was broadly defined as 1 of the following11:
- a Structured Clinical Interview for the DSM (SCID)-rated assessment that revealed the patient had enough symptoms to meet criteria for a DSM-IV major depressive episode
- a 17-item HAM-D scoren of ≥18
- SCID-rated psychosis
- other significant clinical worsening, defined as having a suicide plan or attempting suicide, developing SCID-rated symptoms of mania or hypomania, or being hospitalized in a psychiatric unit.
Compared with sertraline plus placebo, continuing sertraline plus olanzapine reduced the risk of relapse over 36 weeks (hazard ratio, 0.25; 95% confidence interval, 0.13 to 0.48; P < .001).11 However, as expected, the incidence of adverse effects such as weight gain and parkinsonism was higher in the olanzapine group. Therefore, it is important to consider the potential long-term adverse effects of continuing antipsychotic medications.
Weighing the evidence
Electroconvulsive therapy is considered a first-line treatment option for MDD with psychotic features; however, because of limitations associated with this approach, antidepressants plus antipsychotics are often utilized as an initial treatment.
Continue to: CASE
CASE CONTINUED
Based on the results of the STOP-PD and STOP-PD II trials, Mrs. C should be continued on sertraline plus olanzapine for at least another 3 to 6 months before an olanzapine taper should be considered. At that time, the risks and benefits of a taper vs continuing therapy should be considered. Given her history of MDD and the severity of this most recent episode, sertraline therapy should be continued for at least 2 years, and possibly indefinitely.
Related Resources
- Texas Medication Algorithm Project. Algorithm for the treatment of major depressive disorder with psychotic features. https://chsciowa.org/sites/chsciowa.org/files/resource/files/9_-_depression_med_algorithm_supplement.pdf
- Dold M, Bartova L, Kautzky A, et al. Psychotic features in patients with major depressive disorder: a report from the European Group for the Study of Resistant Depression. J Clin Psychiatry. 2019;80(1):17m12090. doi: 10.4088/ JCP.17m12090
- Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7): 622-631.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Sertraline • Zoloft
Venlafaxine • Effexor
Mrs. C, age 56, has a history of major depressive disorder (MDD). She has been stable for 5 years without medication. Six months ago, she presented to you, along with her son, seeking help. She reported that she had been experiencing insomnia, fatigue, and was not engaging in hobbies. Her son told you that his mother had lost weight and had been avoiding family dinners. Mrs. C reported recurrent thoughts of dying and heard voices vividly telling her that she was a burden and that her family would be better off without her. However, there was no imminent danger of self-harm. At that appointment, you initiated
Since that time, Mrs. C has followed up with you monthly with good response to the medications. Currently, she states her depression is much improved, and she denies hearing voices for approximately 5 months.
Based on her presentation and response, what do the data suggest about her length of treatment, and when should you consider tapering the antipsychotic medication?
In DSM-5, MDD with psychotic features is a severe subtype of MDD that is defined as a major depressive episode characterized by delusions and/or hallucinations.1 In the general population, the lifetime prevalence of this disorder varies from 0.35% to 1%, and the rate is higher in older patients.2 Risk factors include female gender, family history, and concomitant bipolar disorder.2
Epidemiologic studies have shown that psychotic features can occur in 15% to 20% of patients with MDD. The psychotic features that occur during these episodes are delusions and hallucinations.1 These features can be either mood-congruent (related to the depressive themes of worthlessness or guilt) or mood-incongruent (ie, unrelated to depressive themes).1
Treatment options: ECT or pharmacotherapy
Guidelines from the American Psychiatric Association3 and the National Institute for Clinical Excellence4 recommend treating depression with psychosis with electroconvulsive therapy (ECT) or with combined antidepressant and antipsychotic medications as first-line options. The Texas Medication Algorithm Project (TMAP) Algorithm for MDD,5 which closely focuses on treatment of MDD with psychotic features, can be used for treatment decisions (see Related Resources).
Electroconvulsive therapy is known to be efficacious in treating patients with MDD with psychotic features and should be considered as a treatment option. However, medication therapy is often chosen as the initial treatment due to the limitations of ECT, including accessibility, cost, and patient preference. However, in certain cases, ECT is the preferred option because it can provide rapid and significant improvement in patients with severe psychosis, suicidality, or risk of imminent harm.
Continue to: Pharmacotherapy
Pharmacotherapy for the treatment of MDD with psychotic features should consist of a combination of an antidepressant and antipsychotic medication. This combination has been shown to be more effective than either agent alone. Some combinations have been studied specifically for MDD with psychosis. The Study of the Pharmacotherapy of Psychotic Depression (STOP-PD), a 12-week, double-blind, randomized controlled trial, found that the combination of sertraline and olanzapine was efficacious and superior to monotherapy with olanzapine in an acute setting.6 In another study, the combination of olanzapine and
How long should treatment last?
The optimal timeline for treating patients with MDD with psychotic features is unknown. According to the TMAP algorithm and expert opinion, the continuation phase of pharmacotherapy should include treatment for at least 4 months with an antipsychotic medication and at least 2 years to lifetime treatment with an antidepressant.5 The STOP-PD II study, which was a continuation of the 12-week STOP-PD study, examined antipsychotic duration to determine the effects of continuing olanzapine once an episode of psychotic depression had responded to olanzapine and sertraline.11 Patients who had achieved remission after receiving olanzapine and sertraline were randomized to continue to receive this combination or to receive sertraline plus placebo for 36 weeks. The primary outcome was relapse, which was broadly defined as 1 of the following11:
- a Structured Clinical Interview for the DSM (SCID)-rated assessment that revealed the patient had enough symptoms to meet criteria for a DSM-IV major depressive episode
- a 17-item HAM-D scoren of ≥18
- SCID-rated psychosis
- other significant clinical worsening, defined as having a suicide plan or attempting suicide, developing SCID-rated symptoms of mania or hypomania, or being hospitalized in a psychiatric unit.
Compared with sertraline plus placebo, continuing sertraline plus olanzapine reduced the risk of relapse over 36 weeks (hazard ratio, 0.25; 95% confidence interval, 0.13 to 0.48; P < .001).11 However, as expected, the incidence of adverse effects such as weight gain and parkinsonism was higher in the olanzapine group. Therefore, it is important to consider the potential long-term adverse effects of continuing antipsychotic medications.
Weighing the evidence
Electroconvulsive therapy is considered a first-line treatment option for MDD with psychotic features; however, because of limitations associated with this approach, antidepressants plus antipsychotics are often utilized as an initial treatment.
Continue to: CASE
CASE CONTINUED
Based on the results of the STOP-PD and STOP-PD II trials, Mrs. C should be continued on sertraline plus olanzapine for at least another 3 to 6 months before an olanzapine taper should be considered. At that time, the risks and benefits of a taper vs continuing therapy should be considered. Given her history of MDD and the severity of this most recent episode, sertraline therapy should be continued for at least 2 years, and possibly indefinitely.
Related Resources
- Texas Medication Algorithm Project. Algorithm for the treatment of major depressive disorder with psychotic features. https://chsciowa.org/sites/chsciowa.org/files/resource/files/9_-_depression_med_algorithm_supplement.pdf
- Dold M, Bartova L, Kautzky A, et al. Psychotic features in patients with major depressive disorder: a report from the European Group for the Study of Resistant Depression. J Clin Psychiatry. 2019;80(1):17m12090. doi: 10.4088/ JCP.17m12090
- Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7): 622-631.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Jääskeläinen E, Juola T, Korpela H, et al. Epidemiology of psychotic depression - systematic review and meta-analysis. Psychol Med. 2018;48(6):905-918.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(4)(suppl):1-45.
4. National Institute for Clinical Excellence. Depression in adults: recognition and management: clinical guideline [CG90]. National Institute for Health and Clinical Excellence. Published October 28, 2009. Accessed January 12, 2021. https://www.nice.org.uk/guidance/cg90
5. Crimson ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry. 1999;60(3):142-156.
6. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy for psychotic depression -- the STOP-PD study. Arch Gen Psychiatry. 2009;66(8):838-847.
7. Rothschild AJ, Williamson DJ, Tohen MF, et al. A double-blind, randomized study of olanzapine and olanzapine/fluoxetine combination for major depression with psychotic features. J Clin Psychopharmacol. 2004;24(4):365-373.
8. Wijkstra J, Burger H, van den Broek WW, et al. Treatment of unipolar psychotic depression: a randomized, doubleblind study comparing imipramine, venlafaxine, and venlafaxine plus quetiapine. Acta Psychiatr Scand. 2010;21(3):190-200.
9. Muller-Siecheneder F, Muller M, Hillert A, et al. Risperidone versus haloperidol and amitriptyline in the treatment of patients with a combined psychotic and depressive syndrome. J Clin Psychopharm. 1998;18(2):111-120.
10. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacological treatment of delusional depression. Am J Psychiatry. 1985;142(4):430-436.
11. Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7):622-631.
1. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013.
2. Jääskeläinen E, Juola T, Korpela H, et al. Epidemiology of psychotic depression - systematic review and meta-analysis. Psychol Med. 2018;48(6):905-918.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(4)(suppl):1-45.
4. National Institute for Clinical Excellence. Depression in adults: recognition and management: clinical guideline [CG90]. National Institute for Health and Clinical Excellence. Published October 28, 2009. Accessed January 12, 2021. https://www.nice.org.uk/guidance/cg90
5. Crimson ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry. 1999;60(3):142-156.
6. Meyers BS, Flint AJ, Rothschild AJ, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy for psychotic depression -- the STOP-PD study. Arch Gen Psychiatry. 2009;66(8):838-847.
7. Rothschild AJ, Williamson DJ, Tohen MF, et al. A double-blind, randomized study of olanzapine and olanzapine/fluoxetine combination for major depression with psychotic features. J Clin Psychopharmacol. 2004;24(4):365-373.
8. Wijkstra J, Burger H, van den Broek WW, et al. Treatment of unipolar psychotic depression: a randomized, doubleblind study comparing imipramine, venlafaxine, and venlafaxine plus quetiapine. Acta Psychiatr Scand. 2010;21(3):190-200.
9. Muller-Siecheneder F, Muller M, Hillert A, et al. Risperidone versus haloperidol and amitriptyline in the treatment of patients with a combined psychotic and depressive syndrome. J Clin Psychopharm. 1998;18(2):111-120.
10. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacological treatment of delusional depression. Am J Psychiatry. 1985;142(4):430-436.
11. Flint AJ, Meyers BS, Rothschild AJ, et al. Effect of continuing olanzapine vs placebo on relapse among patients with psychotic depression in remission: the STOP-PD II randomized clinical trial. JAMA. 2019;322(7):622-631.

Antipsychotics for obsessive-compulsive disorder: Weighing risks vs benefits
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of

Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
When to adjust the dosing of psychotropics in patients with renal impairment
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
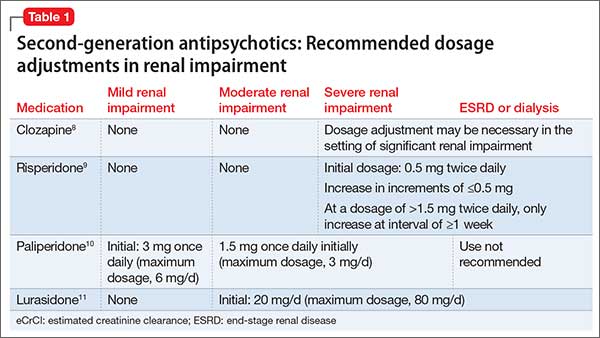
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.