User login
How Should a Patient with Pulmonary Hypertension Be Evaluated, Managed?
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
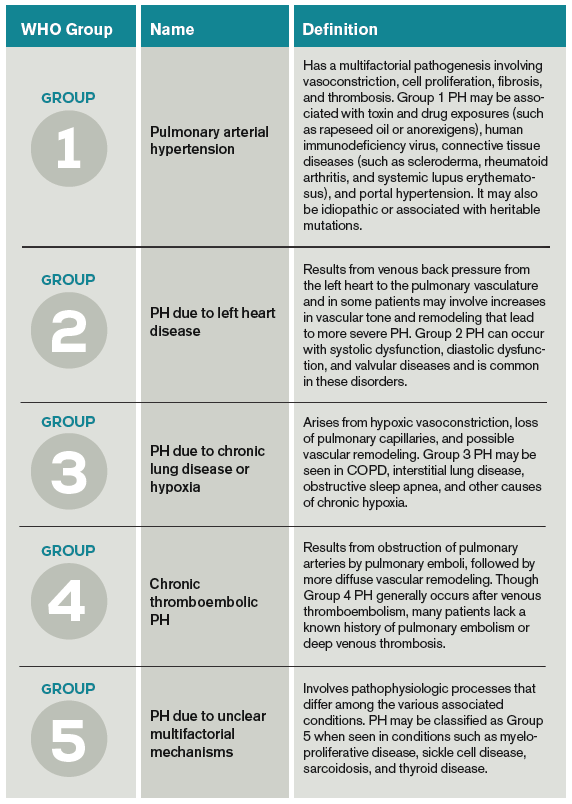
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
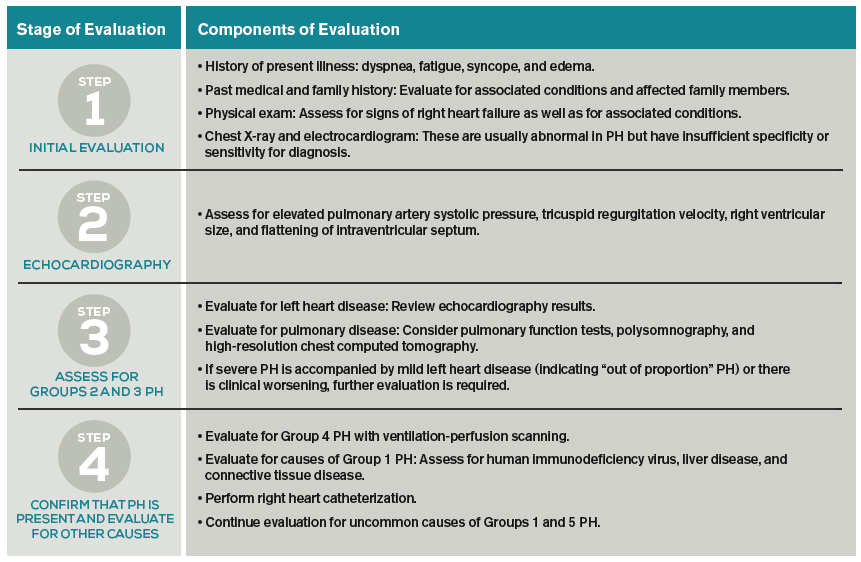
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.

All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.
All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.
All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.