User login
Brodalumab approved for psoriasis with REMS required
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
Minerals used in dermatology part of NIH-Smithsonian exhibit
A geologic display currently on exhibit at the National Institutes of Health Clinical Center in Bethesda, Md., includes several samples of minerals that can be utilized in dermatology.
The “Minerals in Medicine” exhibit, put on by the Clinical Center in partnership with the Smithsonian National Museum of Natural History, includes “more than 40 minerals that are crucial to human health and biomedicine,” according to an NIH statement.
Edward W. Cowen, MD, senior clinician and head of the dermatology consultation service in the National Cancer Institute’s Center for Cancer Research, noted that copper ions have been used for centuries as a disinfecting agent. “In modern medicine, copper-impregnated wound care dressings have been proposed as a mechanism to decrease bacterial colonization with less bacterial resistance than is seen with conventional antibiotic therapy,” he said in an interview.
Sulphur with calcite also is displayed. “Sulfur is found in oral antibiotics used every day, such as penicillin and sulfamethoxazole/trimethoprim, and many topical preparations, ranging from soap to creams to shampoos, where it is effective for the treatment of acne, rosacea, seborrheic dermatitis, and scabies infestation,” Dr. Cowen added. “One common adverse effect of topical medications containing elemental sulfur is the unpleasant smell – sulfur compounds are responsible for the unique fragrance of skunks, among other odors. Interestingly, researchers have found that the exquisite human sensitivity of our olfactory receptors to detect the smell of sulfur is due to another element – copper,” he said.
A geologic display currently on exhibit at the National Institutes of Health Clinical Center in Bethesda, Md., includes several samples of minerals that can be utilized in dermatology.
The “Minerals in Medicine” exhibit, put on by the Clinical Center in partnership with the Smithsonian National Museum of Natural History, includes “more than 40 minerals that are crucial to human health and biomedicine,” according to an NIH statement.
Edward W. Cowen, MD, senior clinician and head of the dermatology consultation service in the National Cancer Institute’s Center for Cancer Research, noted that copper ions have been used for centuries as a disinfecting agent. “In modern medicine, copper-impregnated wound care dressings have been proposed as a mechanism to decrease bacterial colonization with less bacterial resistance than is seen with conventional antibiotic therapy,” he said in an interview.
Sulphur with calcite also is displayed. “Sulfur is found in oral antibiotics used every day, such as penicillin and sulfamethoxazole/trimethoprim, and many topical preparations, ranging from soap to creams to shampoos, where it is effective for the treatment of acne, rosacea, seborrheic dermatitis, and scabies infestation,” Dr. Cowen added. “One common adverse effect of topical medications containing elemental sulfur is the unpleasant smell – sulfur compounds are responsible for the unique fragrance of skunks, among other odors. Interestingly, researchers have found that the exquisite human sensitivity of our olfactory receptors to detect the smell of sulfur is due to another element – copper,” he said.
A geologic display currently on exhibit at the National Institutes of Health Clinical Center in Bethesda, Md., includes several samples of minerals that can be utilized in dermatology.
The “Minerals in Medicine” exhibit, put on by the Clinical Center in partnership with the Smithsonian National Museum of Natural History, includes “more than 40 minerals that are crucial to human health and biomedicine,” according to an NIH statement.
Edward W. Cowen, MD, senior clinician and head of the dermatology consultation service in the National Cancer Institute’s Center for Cancer Research, noted that copper ions have been used for centuries as a disinfecting agent. “In modern medicine, copper-impregnated wound care dressings have been proposed as a mechanism to decrease bacterial colonization with less bacterial resistance than is seen with conventional antibiotic therapy,” he said in an interview.
Sulphur with calcite also is displayed. “Sulfur is found in oral antibiotics used every day, such as penicillin and sulfamethoxazole/trimethoprim, and many topical preparations, ranging from soap to creams to shampoos, where it is effective for the treatment of acne, rosacea, seborrheic dermatitis, and scabies infestation,” Dr. Cowen added. “One common adverse effect of topical medications containing elemental sulfur is the unpleasant smell – sulfur compounds are responsible for the unique fragrance of skunks, among other odors. Interestingly, researchers have found that the exquisite human sensitivity of our olfactory receptors to detect the smell of sulfur is due to another element – copper,” he said.
VIDEO: Topical antifungals win with patients
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM SDEF LAS VEGAS SEMINAR

FDA: Etanercept first biologic approved for pediatric psoriasis
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.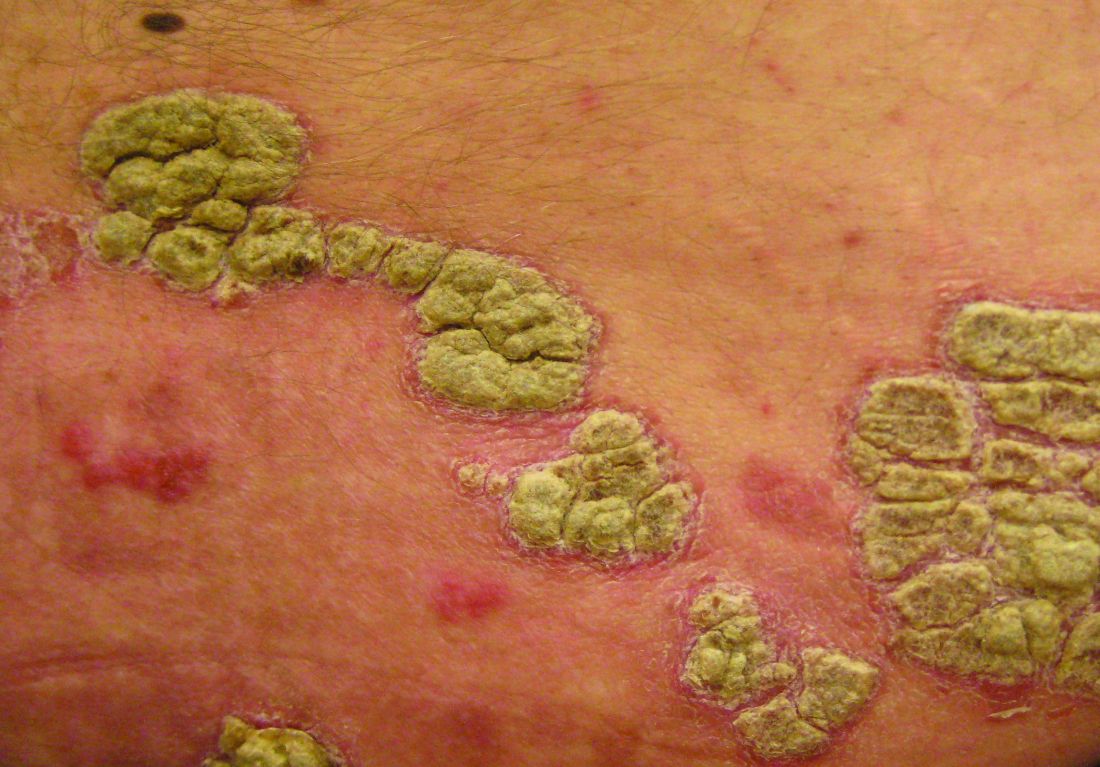
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
FDA accepting comments on draft guidelines on compounding law
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
FDA Approves First Retinoid for OTC Acne Treatment
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
FDA approves first retinoid for OTC acne treatment
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
The Food and Drug Administration has approved adapalene gel 0.1% for over-the-counter use, making it the first retinoid for treating acne that will be available in the United States without a prescription.
The approval also makes the product, marketed as Differin Gel 0.1%, “the first new active ingredient for acne treatment for OTC use since the 1980s,” according to an FDA statement announcing the approval July 8. Differin Gel is approved for use in people aged 12 years and older.
The switch to OTC status was supported by postmarketing safety data; by consumer studies data, which included a label comprehension study, a self-selection study, and an “actual use” study; and data from a “maximal use” study submitted by the manufacturer.
“Overall, results from the consumer studies showed that consumers can understand the information on the OTC label, appropriately select whether the product is right for them, and use the product appropriately,” according to the FDA statement. “The maximal use trial, a study of absorption of the drug through acne-affected skin when applied daily over a large surface area (face, shoulders, upper back, and chest), demonstrated that absorption is limited, thus supporting safe use of Differin Gel 0.1% by people using it OTC.”
The FDA noted that “some other retinoid drugs have been shown to cause birth defects,” and it advises that women “who are pregnant, planning to become pregnant, or [are] breast-feeding” should ask a doctor before using the product.
“While there have been no adequate and well-controlled studies of Differin Gel 0.1% in pregnant women, there is no specific evidence that Differin Gel 0.1%, when used topically as directed, causes birth defects in humans,” according to the FDA.
Consumers should follow the OTC Drug Facts label “and consult with their health care providers if their symptoms do not improve,” the FDA added.
At an April 2016 meeting, the FDA’s nonprescription drugs advisory committee unanimously voted that the safety of adapalene gel 0.1% for OTC use for treating acne had been “adequately demonstrated.”
Galderma Laboratories markets Differin Gel 0.1%.
FDA: No Oral Ketoconazole for Skin, Nail Fungus
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
FDA: No oral ketoconazole for skin, nail fungus
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
The Food and Drug Administration is warning health care professionals not to prescribe oral ketoconazole for patients with fungal infections of the skin and nails, because of "the risks of serious liver damage, adrenal gland problems, and harmful interactions with other medicines that outweigh its benefit in treating these conditions."
The advisory, issued on May 19, points out that oral ketoconazole (Nizoral) is no longer approved for treating nail or skin fungal infections. Topical forms of ketoconazole have not been associated with liver damage, adrenal problems, or drug interactions, the advisory adds.
"Health care professionals should use ketoconazole tablets only to treat serious fungal infections when no other antifungal therapies are available," according to the FDA. "Skin and nail fungal infections in otherwise healthy persons are not life-threatening, and so the risks associated with oral ketoconazole outweigh the benefits. Other treatment options are available over-the-counter and by prescription, but are also associated with risks that should be weighed against their benefits."
The advisory updates one issued in July 2013 when the drug's label was changed to reflect these safety concerns, including dropping the nail and skin infections from the approved indications. Since then, the FDA has received one report of a patient who died of liver failure associated with oral ketoconazole used to treat nail fungus. Furthermore, a survey of office-based physicians found that in the 18 months ending in June 2015, "skin and nail fungal infections were the only diagnoses cited for the use of oral ketoconazole."
Serious adverse events associated with oral ketoconazole should be reported to the FDA's MedWatch program online or call 800-332-1088.
Phase III Dupilumab Data Show Significant Improvements in Atopic Dermatitis
Treatment with dupilumab resulted in significant clinical improvements in adults with inadequately controlled moderate to-severe atopic dermatitis, in two phase III studies evaluating the biologic agent, according to Regeneron and Sanofi.
The phase III results of the two 16-week studies, SOLO 1 and SOLO 2, in nearly 1,400 adults with baseline Investigator’s Global Assessment (IGA) scores of 3 (moderate disease) or 4 (severe), were announced by Regeneron and Sanofi. The companies are codeveloping dupilumab, which inhibits signaling of interleukin-4 and IL-13, “two key cytokines required for the T helper 2 (Th2) immune response,” according to Regeneron.
In the studies, patients were randomized to treatment with 300 mg subcutaneously of dupilumab once a week or every 2 weeks (after a 600-mg loading dose) or placebo, for 16 weeks.
At 16 weeks, significantly more of those in the two treatment groups achieved clearing or near clearing of skin lesions – a primary endpoint – compared with placebo: In SOLO 1 and SOLO 2, respectively, an IGA score of 0 (clear) or 1 (almost clear) was achieved by 37% and 36% of those treated with 300 mg weekly, and 38% and 36% of those treated every 2 weeks, compared with 10% and 8.5% of those on placebo (P less than .0001).
Improvement from baseline in the Eczema Area and Severity Index (EASI) score in the SOLO 1 and SOLO 2 studies, respectively, were 72% and 69% of those treated with 300 mg weekly and 72% and 67% of those treated every 2 weeks, compared with 38% and 31% of those on placebo (P less than .0001).
The rates of adverse events ranged from 65% to 73% for those on dupilumab, and from 65% to 72% for those on placebo. The rates of serious adverse events were 1%-3% among those on dupilumab and 5%-6% for placebo; serious and severe infections were more common among those on placebo. Compared with placebo, injection site reactions were higher among those on dupilumab (10%-20% vs. 7%-8%). Conjunctivitis was more common among dupilumab-treated patients (7%-12% vs. 2% for placebo). One patient stopped treatment because of conjunctivitis.
The phase III results, which were announced in an April 1 press release, will be presented at a future medical meeting, and the companies plan to file for approval with the Food and Drug Administration in the third quarter of 2016.
Treatment with dupilumab resulted in significant clinical improvements in adults with inadequately controlled moderate to-severe atopic dermatitis, in two phase III studies evaluating the biologic agent, according to Regeneron and Sanofi.
The phase III results of the two 16-week studies, SOLO 1 and SOLO 2, in nearly 1,400 adults with baseline Investigator’s Global Assessment (IGA) scores of 3 (moderate disease) or 4 (severe), were announced by Regeneron and Sanofi. The companies are codeveloping dupilumab, which inhibits signaling of interleukin-4 and IL-13, “two key cytokines required for the T helper 2 (Th2) immune response,” according to Regeneron.
In the studies, patients were randomized to treatment with 300 mg subcutaneously of dupilumab once a week or every 2 weeks (after a 600-mg loading dose) or placebo, for 16 weeks.
At 16 weeks, significantly more of those in the two treatment groups achieved clearing or near clearing of skin lesions – a primary endpoint – compared with placebo: In SOLO 1 and SOLO 2, respectively, an IGA score of 0 (clear) or 1 (almost clear) was achieved by 37% and 36% of those treated with 300 mg weekly, and 38% and 36% of those treated every 2 weeks, compared with 10% and 8.5% of those on placebo (P less than .0001).
Improvement from baseline in the Eczema Area and Severity Index (EASI) score in the SOLO 1 and SOLO 2 studies, respectively, were 72% and 69% of those treated with 300 mg weekly and 72% and 67% of those treated every 2 weeks, compared with 38% and 31% of those on placebo (P less than .0001).
The rates of adverse events ranged from 65% to 73% for those on dupilumab, and from 65% to 72% for those on placebo. The rates of serious adverse events were 1%-3% among those on dupilumab and 5%-6% for placebo; serious and severe infections were more common among those on placebo. Compared with placebo, injection site reactions were higher among those on dupilumab (10%-20% vs. 7%-8%). Conjunctivitis was more common among dupilumab-treated patients (7%-12% vs. 2% for placebo). One patient stopped treatment because of conjunctivitis.
The phase III results, which were announced in an April 1 press release, will be presented at a future medical meeting, and the companies plan to file for approval with the Food and Drug Administration in the third quarter of 2016.
Treatment with dupilumab resulted in significant clinical improvements in adults with inadequately controlled moderate to-severe atopic dermatitis, in two phase III studies evaluating the biologic agent, according to Regeneron and Sanofi.
The phase III results of the two 16-week studies, SOLO 1 and SOLO 2, in nearly 1,400 adults with baseline Investigator’s Global Assessment (IGA) scores of 3 (moderate disease) or 4 (severe), were announced by Regeneron and Sanofi. The companies are codeveloping dupilumab, which inhibits signaling of interleukin-4 and IL-13, “two key cytokines required for the T helper 2 (Th2) immune response,” according to Regeneron.
In the studies, patients were randomized to treatment with 300 mg subcutaneously of dupilumab once a week or every 2 weeks (after a 600-mg loading dose) or placebo, for 16 weeks.
At 16 weeks, significantly more of those in the two treatment groups achieved clearing or near clearing of skin lesions – a primary endpoint – compared with placebo: In SOLO 1 and SOLO 2, respectively, an IGA score of 0 (clear) or 1 (almost clear) was achieved by 37% and 36% of those treated with 300 mg weekly, and 38% and 36% of those treated every 2 weeks, compared with 10% and 8.5% of those on placebo (P less than .0001).
Improvement from baseline in the Eczema Area and Severity Index (EASI) score in the SOLO 1 and SOLO 2 studies, respectively, were 72% and 69% of those treated with 300 mg weekly and 72% and 67% of those treated every 2 weeks, compared with 38% and 31% of those on placebo (P less than .0001).
The rates of adverse events ranged from 65% to 73% for those on dupilumab, and from 65% to 72% for those on placebo. The rates of serious adverse events were 1%-3% among those on dupilumab and 5%-6% for placebo; serious and severe infections were more common among those on placebo. Compared with placebo, injection site reactions were higher among those on dupilumab (10%-20% vs. 7%-8%). Conjunctivitis was more common among dupilumab-treated patients (7%-12% vs. 2% for placebo). One patient stopped treatment because of conjunctivitis.
The phase III results, which were announced in an April 1 press release, will be presented at a future medical meeting, and the companies plan to file for approval with the Food and Drug Administration in the third quarter of 2016.