User login
Clinical Conundrum
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
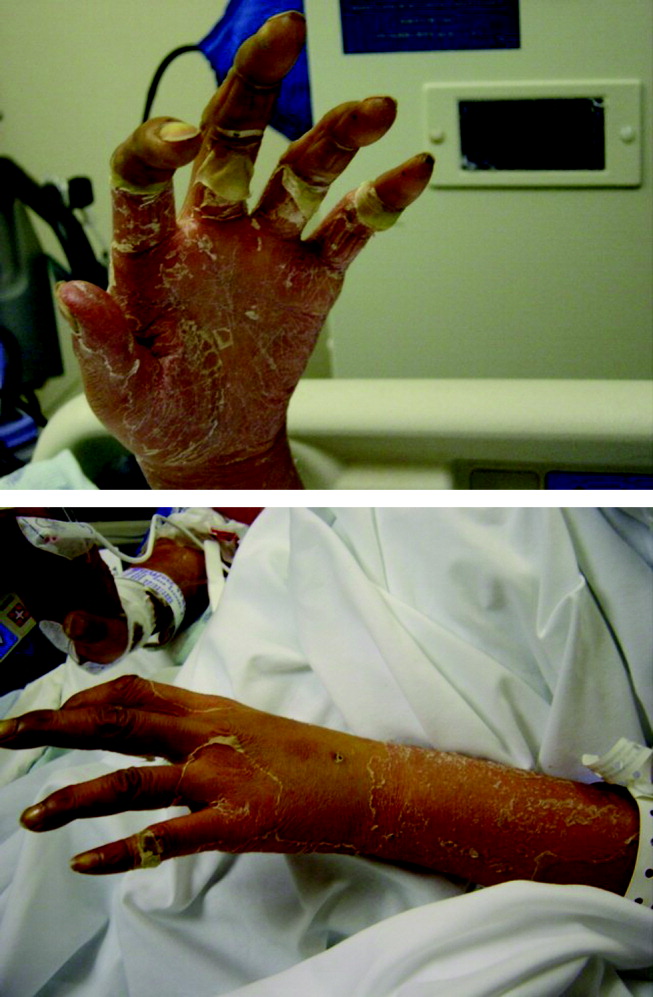
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
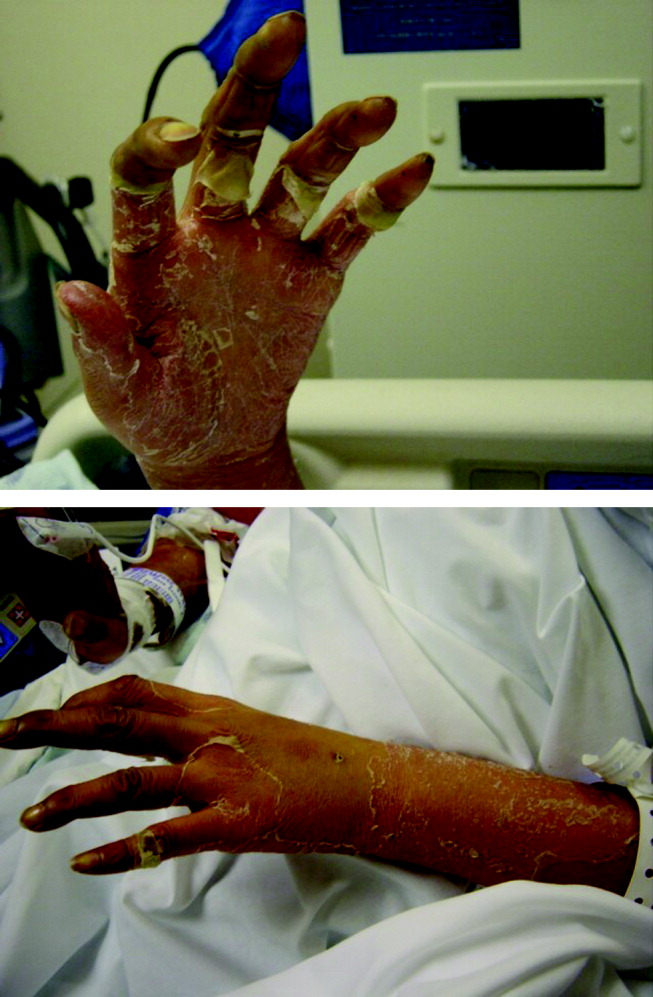
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.

COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.