User login
Hindsight Is 20/20
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.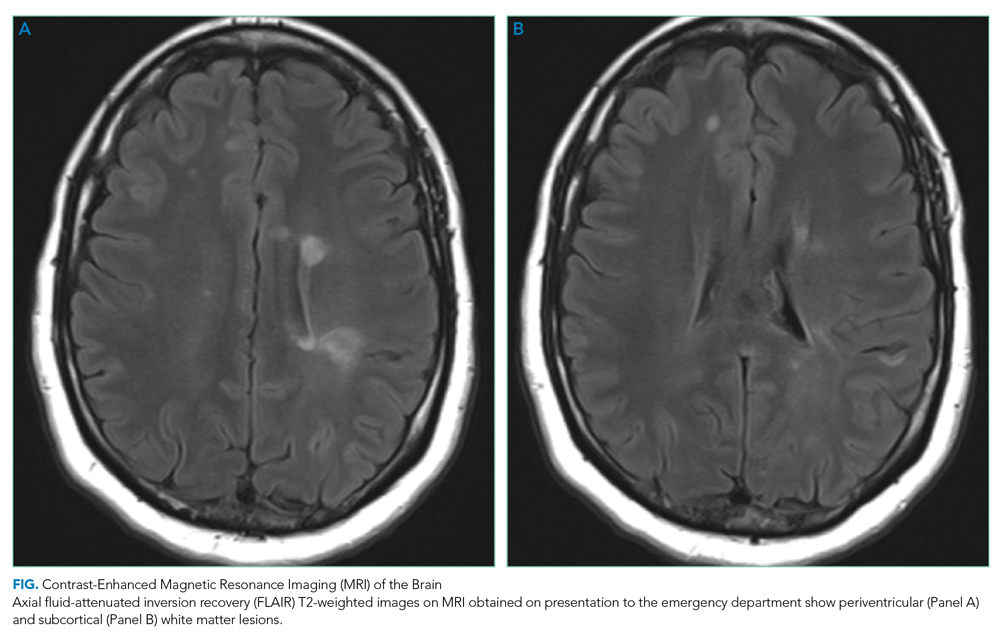
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
A 38-year-old woman presented to her primary care clinic with 3 weeks of progressive numbness and tingling sensation, which began in both hands and then progressed to involv
As with all neurological complaints, localization of the process will often inform a more specific differential diagnosis. If both sensory and motor findings are present, both central and peripheral nerve processes deserve consideration. The onset of paresthesia in the hands, rapid progression to the trunk, and unilateral leg weakness would be inconsistent with a length-dependent peripheral neuropathy. The distribution of complaints and the sacral sparing suggests a myelopathic process involving the cervical region rather than a cauda equina or conus lesions. In an otherwise healthy person of this age and gender, an inflammatory demyelinating disease affecting the cord including multiple sclerosis (MS) would be a strong consideration, although metabolic, vascular, infectious, compressive, or neoplastic disease of the spinal cord could also present with similar subacute onset and pattern of deficits.
Her medical history included morbid obesity, dry eyes, depression, iron deficiency anemia requiring recurrent intravenous replenishment, and abnormal uterine bleeding. Her surgical history included gastric band placement 7 years earlier with removal 5 years later due to persistent gastroesophageal reflux disease, dysphagia, nausea, and vomiting. The gastric band removal was complicated by chronic abdominal pain. Her medications consisted of duloxetine, intermittent iron infusions, artificial tears, loratadine, and pregabalin. She was sexually active with her husband. She consumed alcohol occasionally but did not smoke tobacco or use illicit drugs.
On exam, her temperature was 36.6°C (97.8°F), blood pressure 132/84 mm Hg, and heart rate 85 beats per minute. Body mass index was 39.5 kg/m2. The cardiac, pulmonary, and skin examinations were normal. The abdomen was soft with diffuse tenderness to palpation without rebound or guarding. Examination of cranial nerves 2-12 was normal. Cognition, strength, proprioception, deep tendon reflexes, and light touch were all normal. Her gait was normal, and the Romberg test was negative.
The normal neurologic exam is reassuring but imperfectly sensitive and does not eliminate the possibility of underlying neuropathology. Bariatric surgery may result in an array of nutritional deficiencies such as vitamin E, B12, and copper, which can cause myelopathy and/or neuropathy. However, these abnormalities occur less frequently with gastric banding procedures. If her dry eyes are part of the sicca syndrome, an underlying autoimmune diathesis may be present. Her unexplained chronic abdominal pain prompts considering nonmenstrual causes of iron deficiency anemia, such as celiac disease. Bariatric surgery may contribute to iron deficiency through impaired iron absorption. Her stable weight and lack of diarrhea argue against Crohn’s or celiac disease. Iron deficiency predisposes individuals to pica, most commonly described with ice chip ingestion. If lead pica had occurred, abdominal and neurological symptoms could result. Nevertheless, the abdominal pain is nonspecific, and its occurrence after gastric band removal makes its link to her neurologic syndrome unclear. An initial evaluation would include basic metabolic panel, complete blood count with differential, erythrocyte sedimentation rate, C-reactive protein (CRP), thyroid-stimulating hormone, vitamin B12, and copper levels.
A basic metabolic panel was normal. The white cell count was 5,710 per cubic millimeter, hemoglobin level 12.2 g per deciliter, mean corpuscular volume 85.2 fl, and platelet count 279,000 per cubic millimeter. The serum ferritin level was 18 ng per milliliter (normal range, 13-150), iron 28 µg per deciliter (normal range, 50-170), total iron-binding capacity 364 µg per deciliter (normal range, 250-450), and iron saturation 8% (normal range, 20-55). The vitamin B12 level was 621 pg per milliliter (normal range, 232-1,245) and thyroid-stimulating hormone level 1.87 units per milliliter (normal range, 0.50-4.50). Electrolyte and aminotransferase levels were within normal limits. CRP was 1.0 mg per deciliter (normal range, <0.5) and erythrocyte sedimentation rate 33 millimeters per hour (normal range, 4-25). Hepatitis C and HIV antibodies were nonreactive.
The ongoing iron deficiency despite parenteral iron replacement raises the question of ongoing gastrointestinal or genitourinary blood loss. While the level of vitamin B12 in the serum may be misleadingly normal with cobalamin deficiency, a methylmalonic acid level is indicated to evaluate whether tissue stores are depleted. Copper levels are warranted given the prior bariatric surgery. The mild elevations of inflammatory markers are nonspecific but reduce the likelihood of a highly inflammatory process to account for the neurological and abdominal symptoms.
At her 3-month follow-up visit, she noted that the paresthesia had improved and was now limited to her bilateral lower extremities. During the same clinic visit, she experienced a 45-minute episode of ascending left upper extremity numbness. Her physical examination revealed normal strength and reflexes. She had diminished response to pinprick in both legs to the knees and in both hands to the wrists. Vibration sense was diminished in the bilateral lower extremities.
A glycosylated hemoglobin (HbA1c) level was 6.2%. Methylmalonic acid was 69 nmol per liter (normal range, 45-325). Antibodies to Borrelia burgdorferi and Treponema pallidum were absent. Impaired glucose metabolism was the leading diagnosis for her polyneuropathy, and it was recommended that she undergo an oral glucose tolerance test. Electromyography was not performed.
The neurological symptoms are now chronic, and importantly, the patient has developed sensory deficits on neurological examination, suggesting worsening of the underlying process. While the paresthesia is now limited to a “stocking/glove” distribution consistent with distal sensory polyneuropathy, there should still be a concern for spinal cord pathology given that the HbA1c level of 6.2 would not explain her initial distribution of symptoms. Myelopathy may mimic peripheral nerve disease if, for example, there is involvement of the dorsal columns leading to sensory deficits of vibration and proprioception. Additionally, the transient episode of upper extremity numbness raises the question of sensory nerve root involvement (ie, sensory radiculopathy). Unexplained abdominal pain could possibly represent the involvement of other nerve roots innervating the abdominal wall. The patient’s episode of focal arm numbness recalls the lancinating radicular pain of tabes dorsalis; however, the negative specific treponemal antibody test excludes neurosyphilis.
The differential diagnosis going forward will be strongly conditioned by the localization of the neurological lesion(s). To differentiate between myelopathy, radiculopathy, and peripheral neuropathy, I would perform nerve conduction studies, magnetic resonance imaging (MRI) of the spinal cord, and cerebrospinal fluid analysis.
The patient began taking a multivitamin, and after weeks her paresthesia had resolved. One month later, she developed an intermittent, throbbing left-sided headache and pain behind the left eye that was worsened with ocular movement. She then noted decreased visual acuity in her left eye that progressed the following month. She denied photophobia, flashers, or floaters.
In the emergency department, visual acuity was 20/25 in her right eye; in the left eye she was only able to count fingers. Extraocular movements of both eyes were normal as was her right pupillary reflex. Red desaturation and a relative afferent papillary defect were present in the left eye. Fundoscopic exam demonstrated left optic disc swelling. The remainder of her cranial nerves were normal. She had pronation of the left upper extremity and mild right finger-to-nose dysmetria. Muscle tone, strength, sensation, and deep tendon reflexes were normal.
The improvement in the sensory symptoms was unlikely to be related to the nutritional intervention and provides a clue to an underlying waxing and waning illness. That interpretation is supported by the subsequent development of new visual symptoms and signs, which point to optic nerve pathology. Optic neuropathy has a broad differential diagnosis that includes ischemic, metabolic, toxic, and compressive causes. Eye pain, swelling of the optic disc, and prominent impairment of color vision all point to the more specific syndrome of optic neuritis caused by infections (including both Treponema pallidum and Borrelia species), systemic autoimmune diseases (systemic lupus erythematosus or Sjogren’s syndrome), and central nervous system (CNS) demyelinating diseases. Of these, inflammatory demyelinating processes would be the likeliest explanation of intermittent and improving neurologic findings.
With relapsing symptoms and findings that are separate in distribution and time, two diagnoses become most likely, and both of these are most often diagnosed in young women. MS is common, and optic neuritis occurs in more than 50% of patients over the course of illness. Neuromyelitis optica spectrum disorder (NMOSD) is a rare condition that can exist in isolation or be associated with other autoimmune illnesses. While these entities are difficult to differentiate clinically, neuroimaging that demonstrates extensive intracerebral demyelinating lesions and cerebrospinal fluid with oligoclonal bands favor MS, whereas extensive, predominant spinal cord involvement is suggestive of NMOSD. Approximately 70% of NMO patients harbor an antibody directed against the aquaporin-4 channel, and these antibodies are not seen in patients with MS. A milder NMO-like disorder has also been associated with antimyelin oligodendrocyte antibodies (MOG).
Testing for antinuclear antibodies, anti–double-stranded DNA, anti-Ro (SSA), and anti-La (SSB) antibodies was negative. The level of C3 was 162 mg per deciliter (normal range 81-157) and C4 38 (normal range 13-39). T-spot testing for latent tuberculosis was negative.
There is no serological evidence of active systemic lupus erythematosus or Sjogren’s syndrome. The pretest probability of CNS tuberculosis was low in light of her presenting complaints, relatively protracted course, and overall clinical stability without antituberculous therapy. Tests for latent tuberculosis infection have significant limitations of both sensitivity and specificity for the diagnosis of active disease.
Optical coherence tomography showed optic disc edema in the left eye only. MRI of the head with contrast revealed abnormal signal intensity involving the posterior aspect of the pons, right middle cerebellar peduncle, anterior left temporal lobe, bilateral periventricular white matter, subcortical white matter of the frontal lobes bilaterally, and medulla with abnormal signal and enhancement of the left optic nerve (Figure, Panel A). MRI of the cervical and thoracic spine demonstrated multifocal demyelinating lesions at C3, C4, C7, T4, T5, T7, and T8 (Figure, Panel B). The lesions were not longitudinally extensive. There was no significant postcontrast enhancement to suggest active demyelination.
The cerebrospinal fluid analysis revealed glucose of 105 mg per deciliter and a total protein of 26.1 mg per deciliter. In the fourth tube, there were 20 red cells per cubic and four white cells with a differential of 62% neutrophils, 35% lymphocytes, and 3% monocytes. Epstein-Barr and herpes simplex virus DNA were negative. A Venereal Disease Research Laboratory test was negative. Multiple oligoclonal IgG bands were identified only in the cerebrospinal fluid. Aquaporin-4 IgG and MOG antibodies were negative.
In addition to the expected finding of enhancement of the optic nerve, MRI demonstrated numerous multifocal white matter lesions throughout the cerebrum, brainstem, and spinal cord. Many of the lesions were in “silent” areas, which is not directly attributable to specific symptoms, but several did correlate with the subtler deficits of weakness and dysmetria that were noted on examination. Although such lesions may be seen with a diverse group of systemic diseases including adrenal leukodystrophy, sarcoidosis, Behcet’s, cerebral lupus, and vasculitis, primary CNS inflammatory demyelinating diseases are much more likely. The extensive distribution of demyelination argues against NMOSD. The negative aquaporin-4 and MOG assays support this conclusion. Not all multifocal CNS demyelination is caused by MS and can be seen in posterior reversible encephalopathy syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and adult polyglucosan body disease. Osmotic demyelination is increasingly being recognized as a process that can be more widespread rather than just being limited to the pons. Viral infections of the CNS such as the JC virus (PML) may also provoke multifocal demyelination. Acute disseminated encephalomyelitis is most often seen during childhood, usually after vaccination or after an infectious prodrome. The tempo of the progression of these other diseases tends to be much more rapid than this woman’s course, and often, the neurological deficits are more profound and debilitating. The clinical presentation of sensory-predominant myelopathy, followed by optic neuritis, absence of systemic inflammatory signs or laboratory markers, exclusion of other relevant diseases, multifocal white matter lesions on imaging, minimal pleocytosis, and presence of oligoclonal bands in cerebrospinal fluid, all point to a diagnosis of relapsing-remitting MS.
The patient was diagnosed with MS. She was admitted to the neurology service and treated with 1,000 mg IV methylprednisolone for 3 days with a prompt improvement in her vision. She was started on natalizumab without a relapse of symptoms over the past year.
COMMENTARY
Multiple sclerosis is a chronic demyelinating disease of the CNS.1 The diagnosis of MS has classically been based upon compatible clinical and radiographic evidence of pathology that is disseminated in space and time. Patients typically present with an initial clinically isolated syndrome—involving changes in vision, sensation, strength, mobility, or cognition—for which there is radiographic evidence of demyelination.2 A diagnosis of clinically definite MS is then often made based on a subsequent relapse of symptoms.3
An interval from initial symptoms has been central to the diagnosis of MS (“lesions disseminated in time”). However, recent evidence questions this diagnostic paradigm, and a more rapid diagnosis of MS has been recommended. This recommendation is reflected in the updated McDonald criteria, according to which, if a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.4 The importance of such early diagnosis has been supported by numerous studies that have demonstrated improved clinical outcomes with early therapy.5-7
Despite the McDonald criteria, delays in definitive diagnosis are common in MS. Patients with MS in Spain were found to experience a 2-year delay from the first onset of symptoms to diagnosis.8 In this cohort, patients exhibited delays in presenting to a healthcare provider, as well as delays in diagnosis with an average time from seeing an initial provider to diagnosis of 6 months. When patients who were referred for a demyelinating episode were surveyed, over a third reported a prior suggestive event.9 The time from the first suggestive episode to referral to a neurologist for a recognized demyelinating event was 46 months. Other studies have shown that delays in diagnosis are especially common in younger patients, those with primary progressive MS, and those with comorbid disease.10,11
Misapplication of an MS diagnosis also occurs frequently. In one case series, such misapplication was found most often in cases involving migraine, fibromyalgia, psychogenic disorders, and NMOSD.12 NMOSD is distinguished from MS by the presence of typical brain and spine findings on MRI.13 Antibodies to aquaporin-4 are highly specific and moderately sensitive for the disease.14 It is important to distinguish NMOSD from MS as certain disease-modifying drugs used for MS might actually exacerbate NMOSD.15 A lesion that traverses over three or more contiguous vertebral segments with predominant involvement of central gray matter (ie, longitudinally extensive transverse myelitis) on MRI is the most distinct finding of NMOSD. In contrast, similar to our patient, short and often multiple lesions are demonstrated on spinal cord MRI in patients with MS. Sensitive and specific findings of brain MRI in patients with MS include the presence of lateral ventricle and inferior temporal lobe lesion, Dawson’s fingers, central vein sign, or an S-shaped U-fiber lesion. In NMOSD, brain MRI might reveal periependymal lesions surrounding the ventricular system.
This case highlights the diagnostic challenges related to presentations of a waxing and waning neurological process. At the time of the second evaluation, the presentation was interpreted as a length-dependent polyneuropathy due to glucose intolerance. Our patient’s relatively normal HbA1c, subacute onset of neuropathic symptoms (ie, <4 weeks), sensory and motor complaints, and onset in the upper extremities suggested an alternative diagnosis to prediabetes. Once the patient presented with optic neuritis, the cause of the initial symptoms was obvious, but then, hindsight is 20/20.
TEACHING POINTS
- Early treatment of MS results in improved clinical outcomes.
- Delays in the definitive diagnosis of MS are common, especially in younger patients, those with primary progressive MS, and those with comorbid disease.
- If a clinical presentation is supported by the presence of oligoclonal bands in the cerebrospinal fluid, a diagnosis of MS can be made on the basis of radiographic evidence of dissemination of disease in space, without evidence of dissemination in time.
Acknowledgments
The authors wish to thank Rabih Geha, MD, and Gurpreet Dhaliwal, MD, for providing feedback on an earlier version of this manuscript.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-180. https://doi.org/10.1056/NEJMra140148.
2. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389(10076):1336-1346. https://doi.org/10.1016/S0140-6736(16)30959-X.
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. https://doi.org/10.1016/S0140-6736(18)30481-1.
4. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. https://doi.org/10.1016/S1474-4422(17)30470-2.
5. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389(10076):1347-1356. https://doi.org/10.1016/S0140-6736(16)32388-1.
6. Freedman MS, Comi G, De Stefano N, et al. Moving toward earlier treatment of multiple sclerosis: Findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014;3(2):147-155. https://doi.org/10.1016/j.msard.2013.07.001.
7. Harding K, Williams O, Willis M, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76(5):536-541. https://doi.org/10.1001/jamaneurol.2018.4905.
8. Fernández O, Fernández V, Arbizu T, et al. Characteristics of multiple sclerosis at onset and delay of diagnosis and treatment in Spain (the Novo Study). J Neurol. 257(9):1500-1507. https://doi.org/10.1007/s00415-010-5560-1.
9. Gout O, Lebrun-Frenay C, Labauge P, et al. Prior suggestive symptoms in one-third of patients consulting for a “first” demyelinating event. J Neurol Neurosurg Psychiatry 2011;82(3):323-325. https://doi.org/10.1136/jnnp.2008.166421.
10. Kingwell E, Leung A, Roger E, et al. Factors associated with delay to medical recognition in two Canadian multiple sclerosis cohorts. J Neurol Sci. 2010(1-2);292:57-62. https://doi.org/10.1016/j.jns.2010.02.007.
11. Marrie RA, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity delays diagnosis and increases disability at diagnosis in MS. Neurology. 2009;72(2):117-124. https://doi.org/10.1212/01.wnl.0000333252.78173.5f.
12. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399. https://doi.org/10.1212/WNL.0000000000003152.
13. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology. 2015;84(11):1165-1173. https://doi.org/10.1212/WNL.0000000000001367.
14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. https://doi.org/10.1212/WNL.0000000000001729.
15. Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-1066. https://doi.org/10.1212/WNL.0b013e31826845fe.
© 2020 Society of Hospital Medicine
Cut to the Quick
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
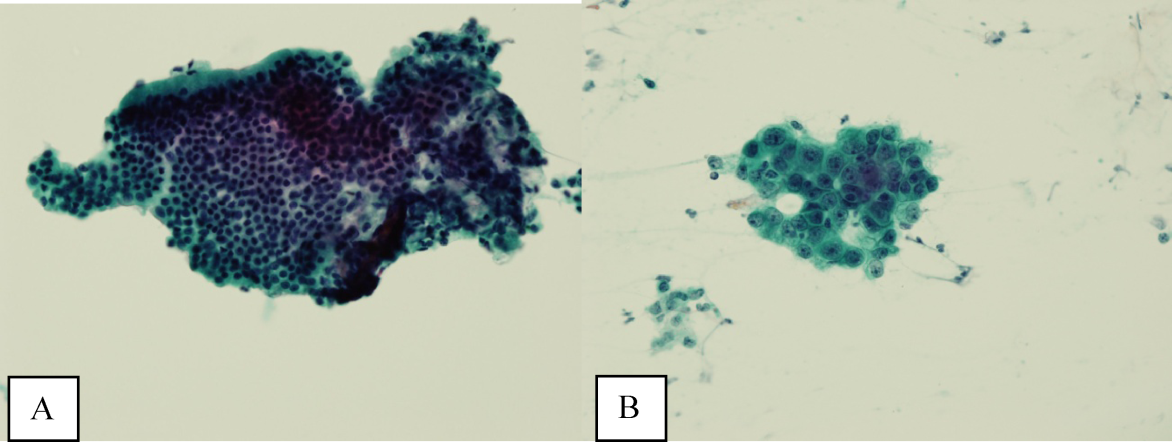
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
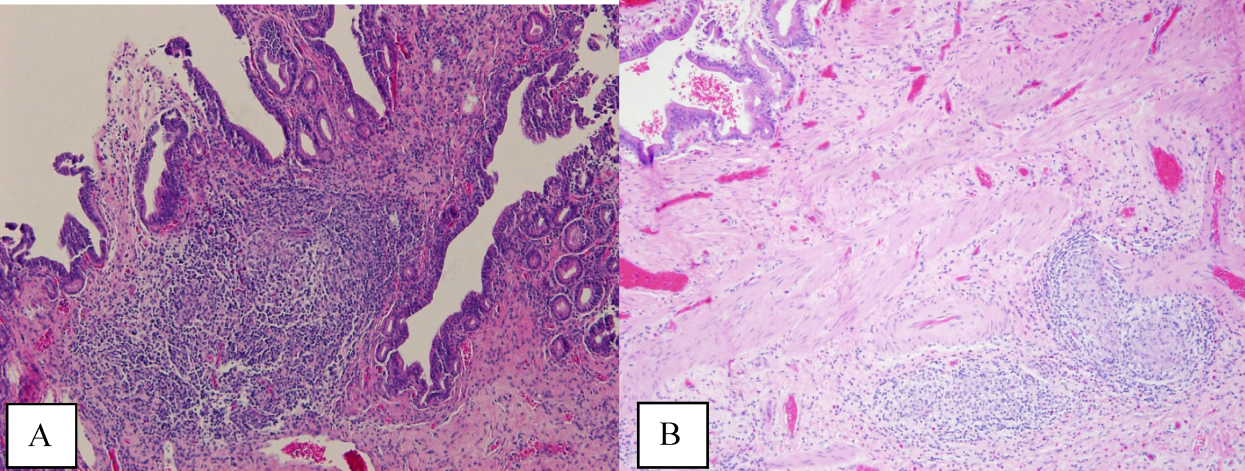
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
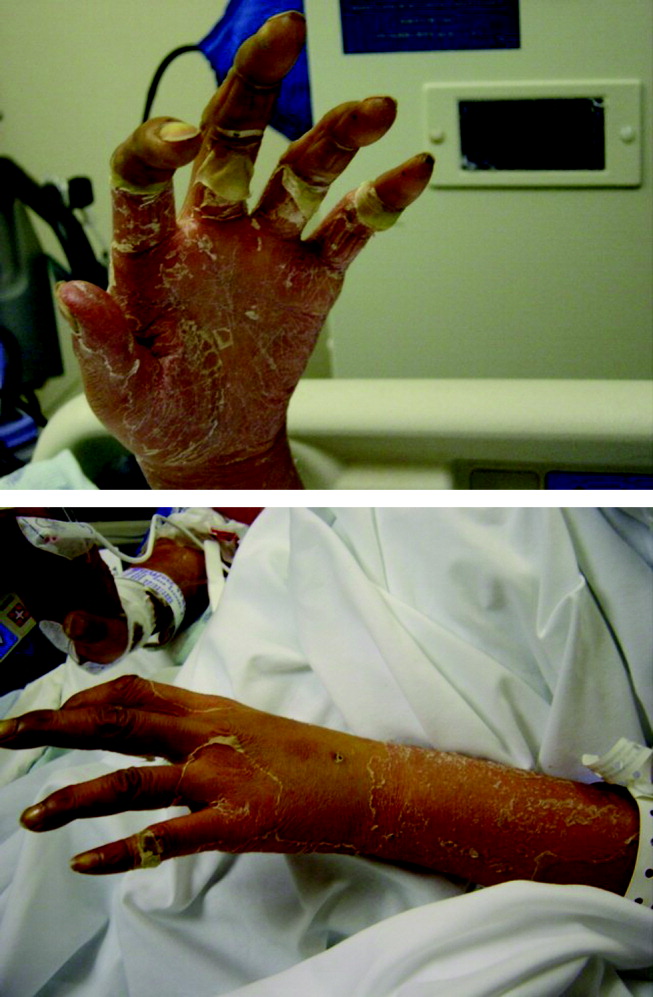
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at <2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at <2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at <2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
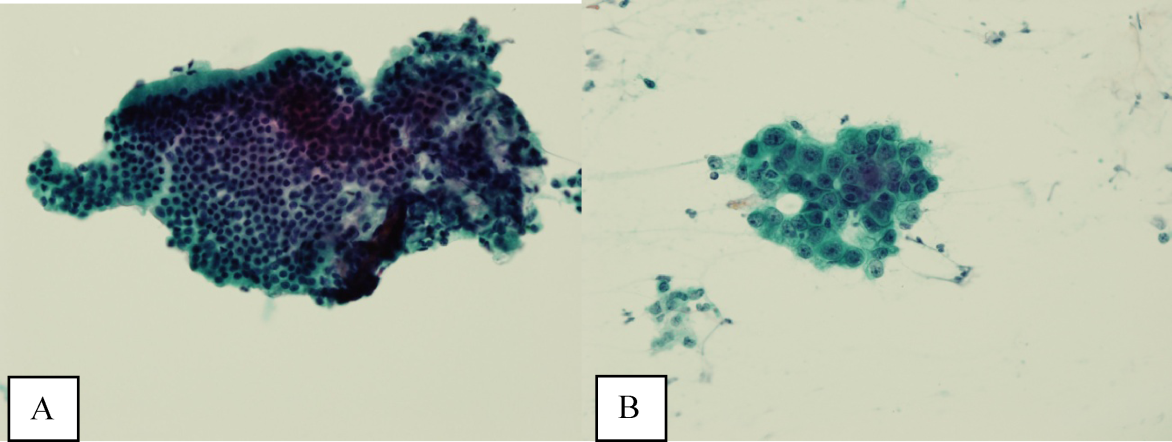
The Hand That Feeds You
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
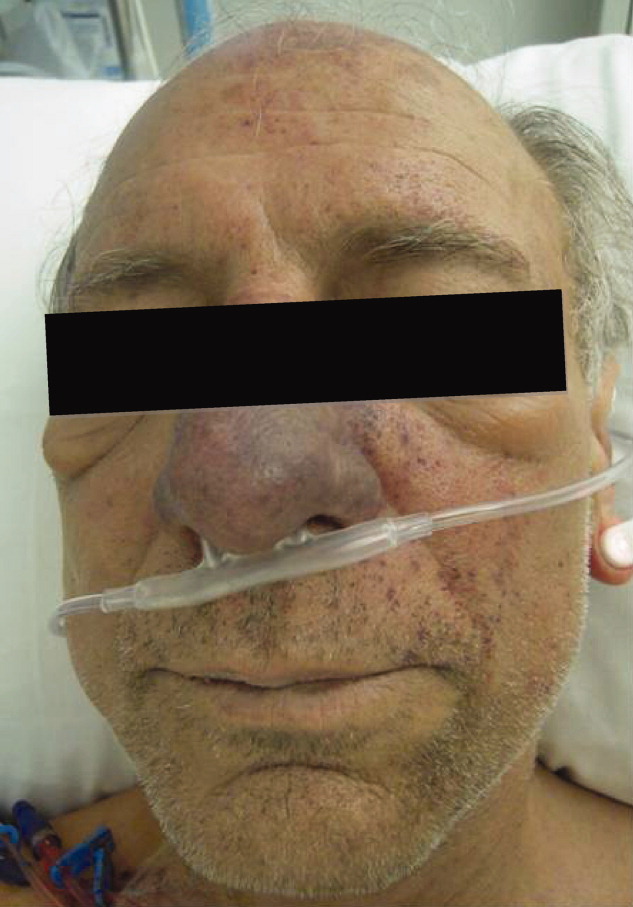
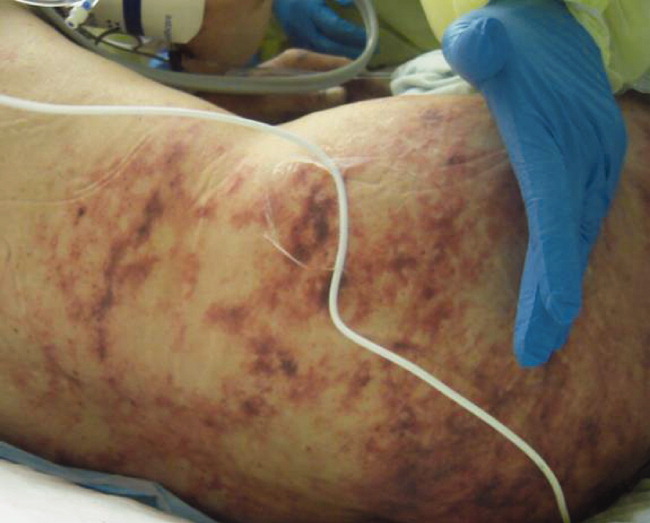
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
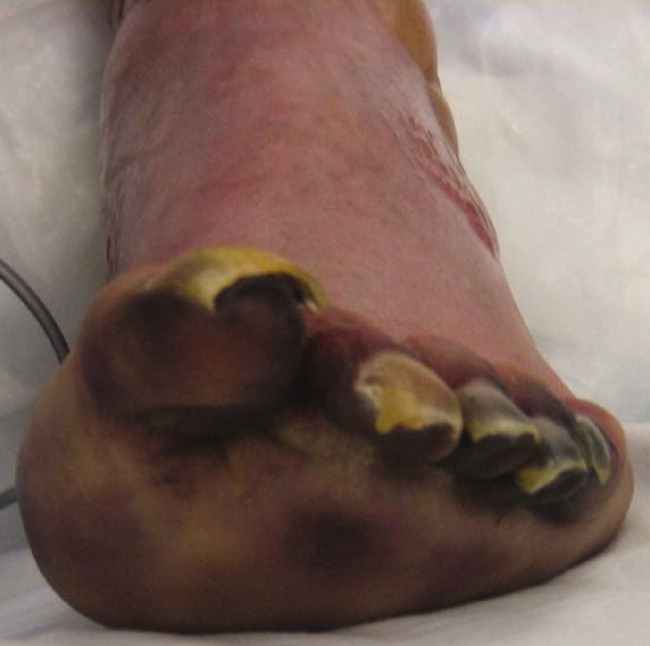
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
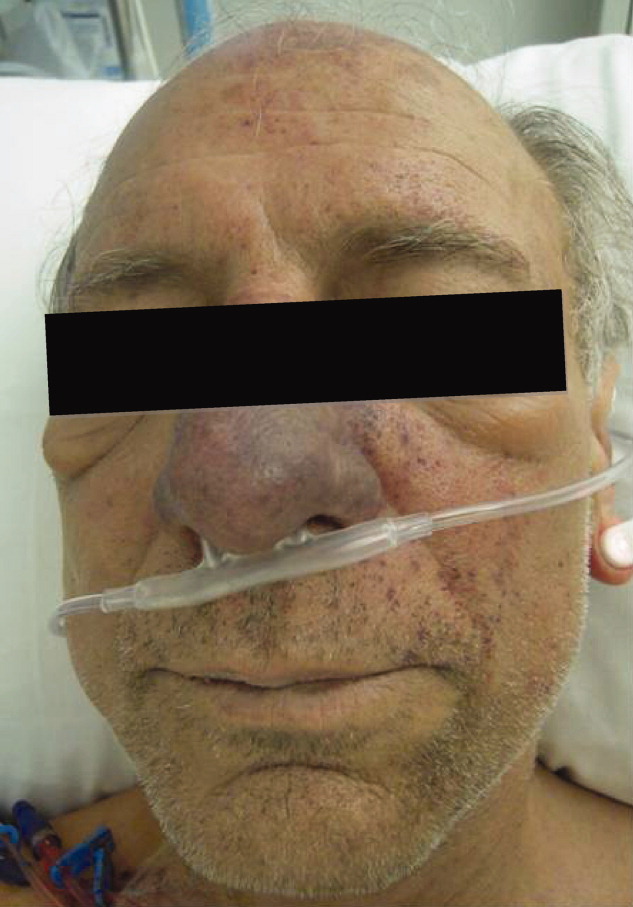
Clinical Conundrum
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
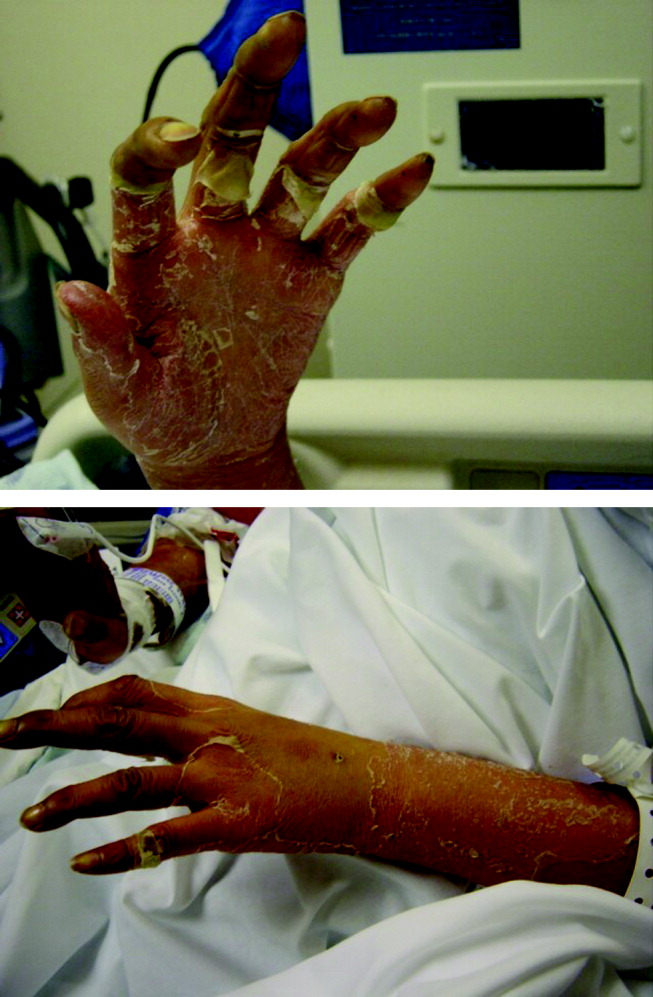
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.

COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets < 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.