User login
Unsuspected Lymphomatoid Granulomatosis in a Patient With Antisynthetase Syndrome
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
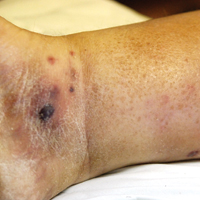
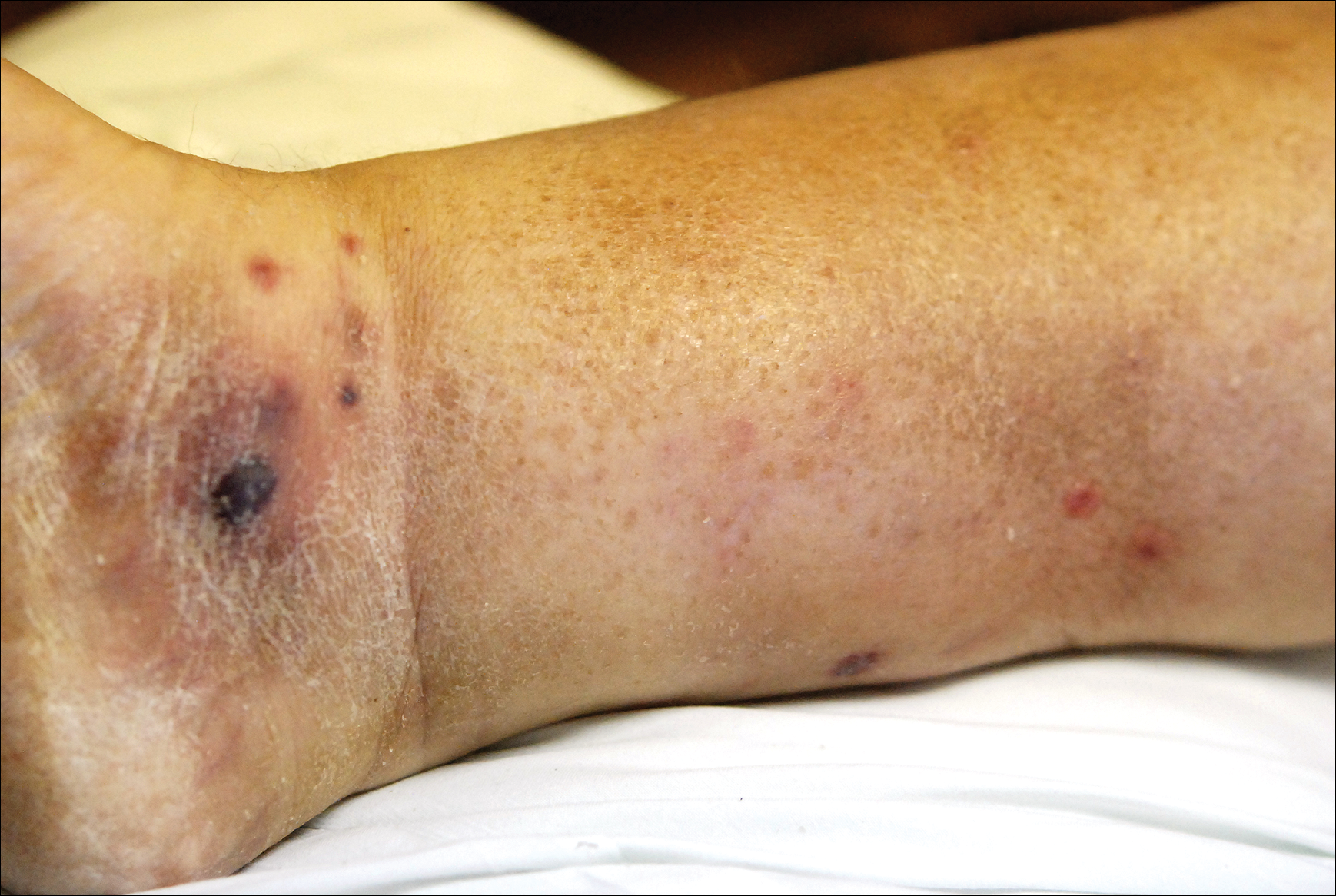
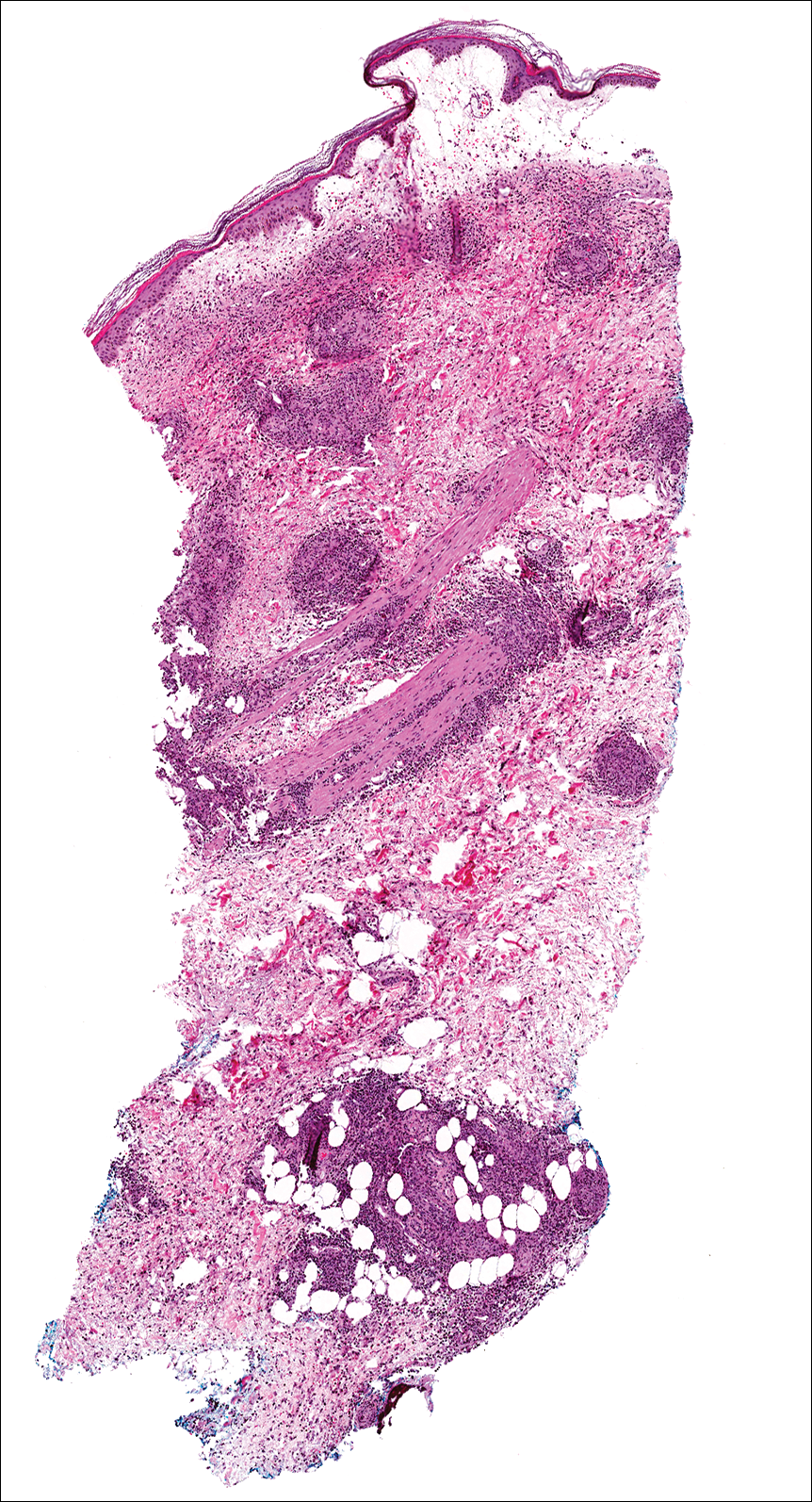
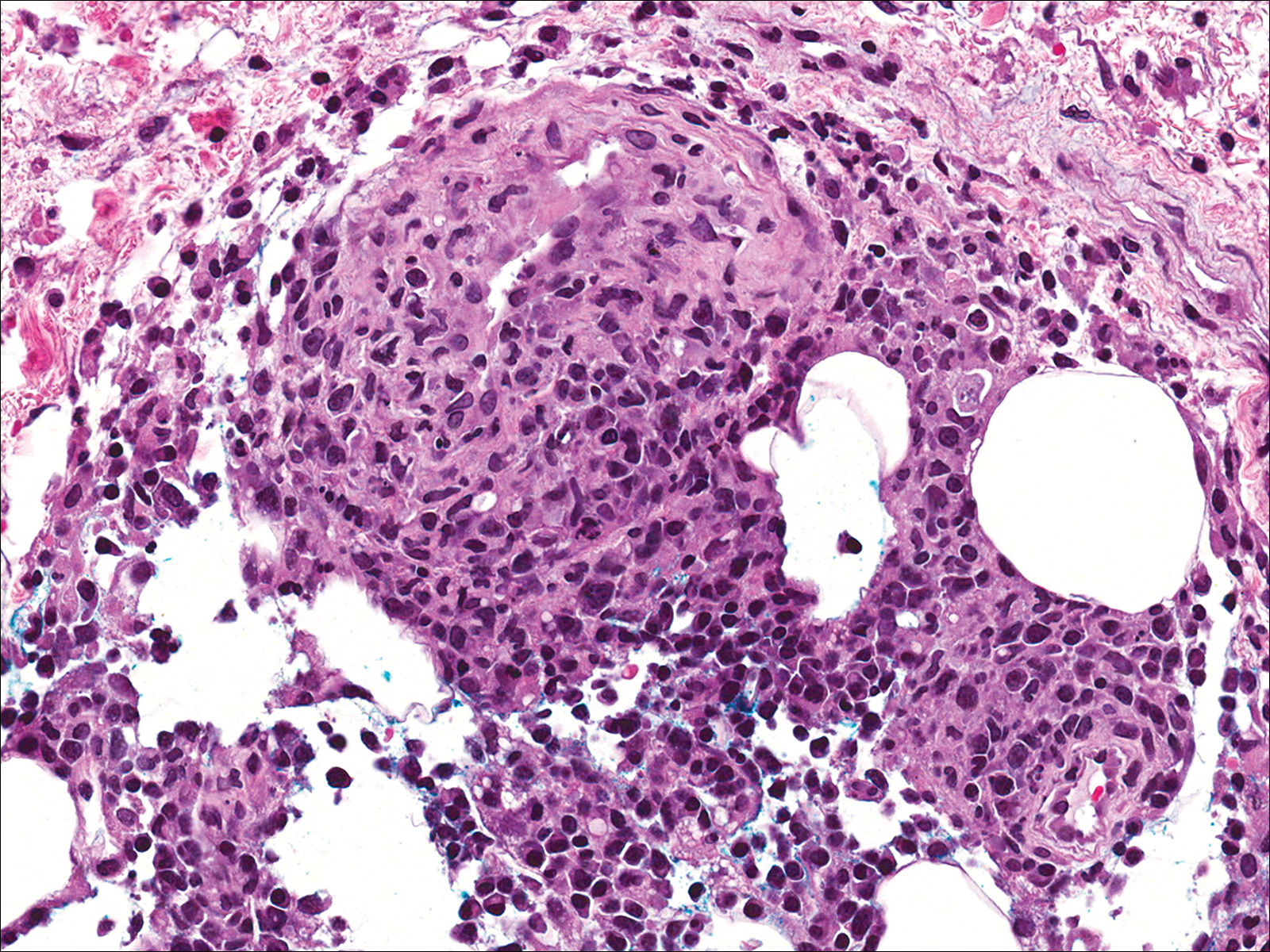
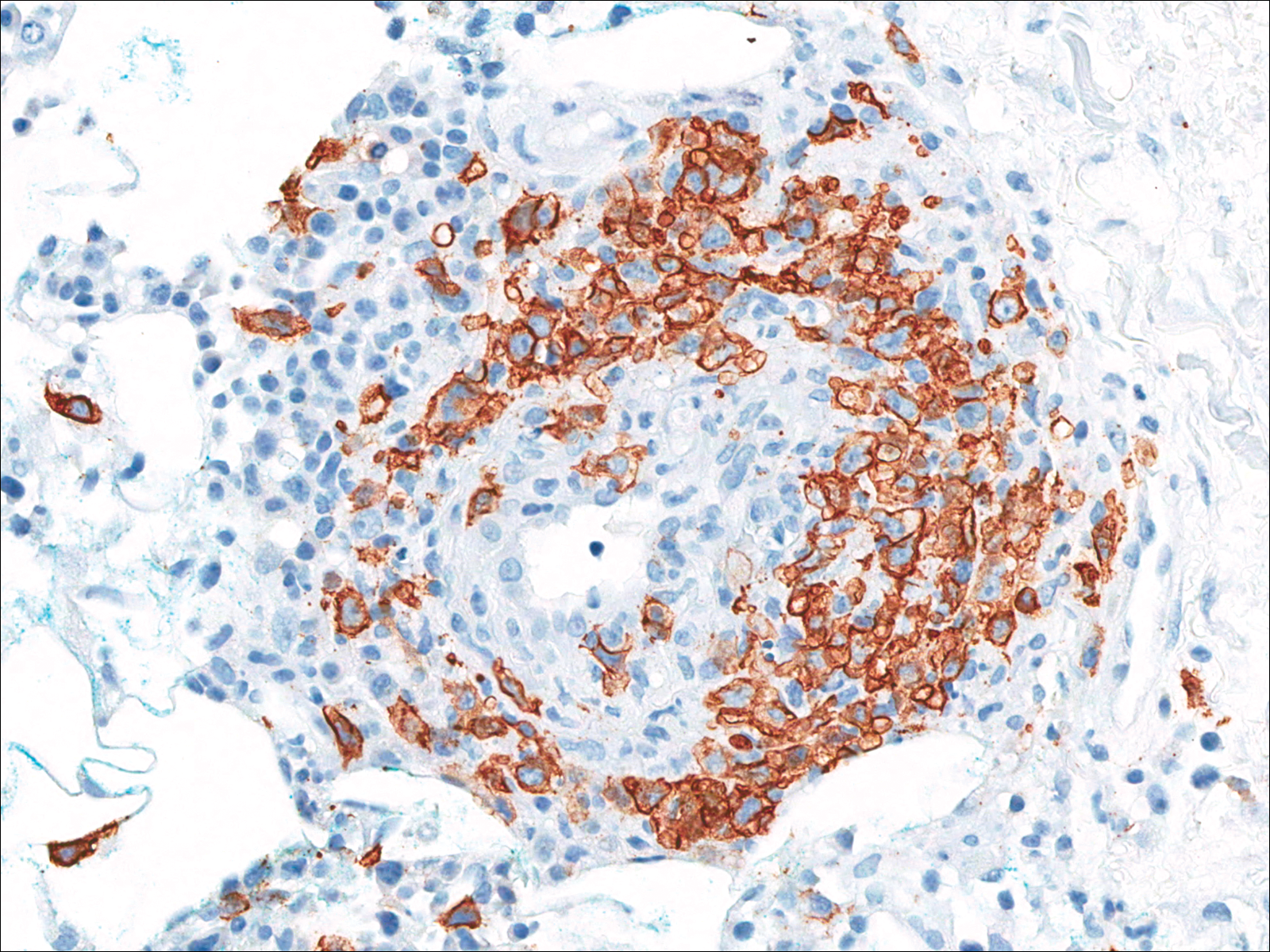
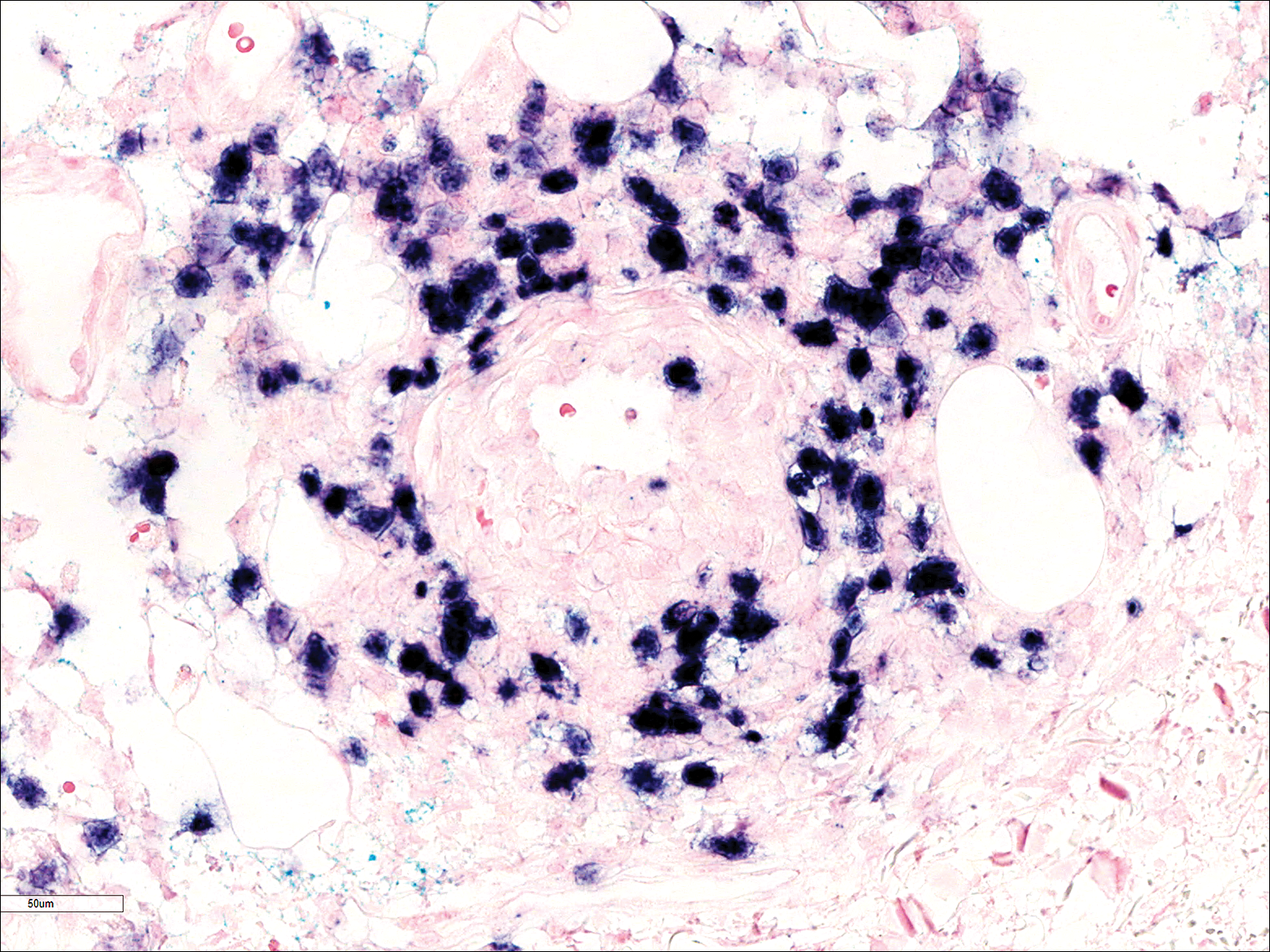
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
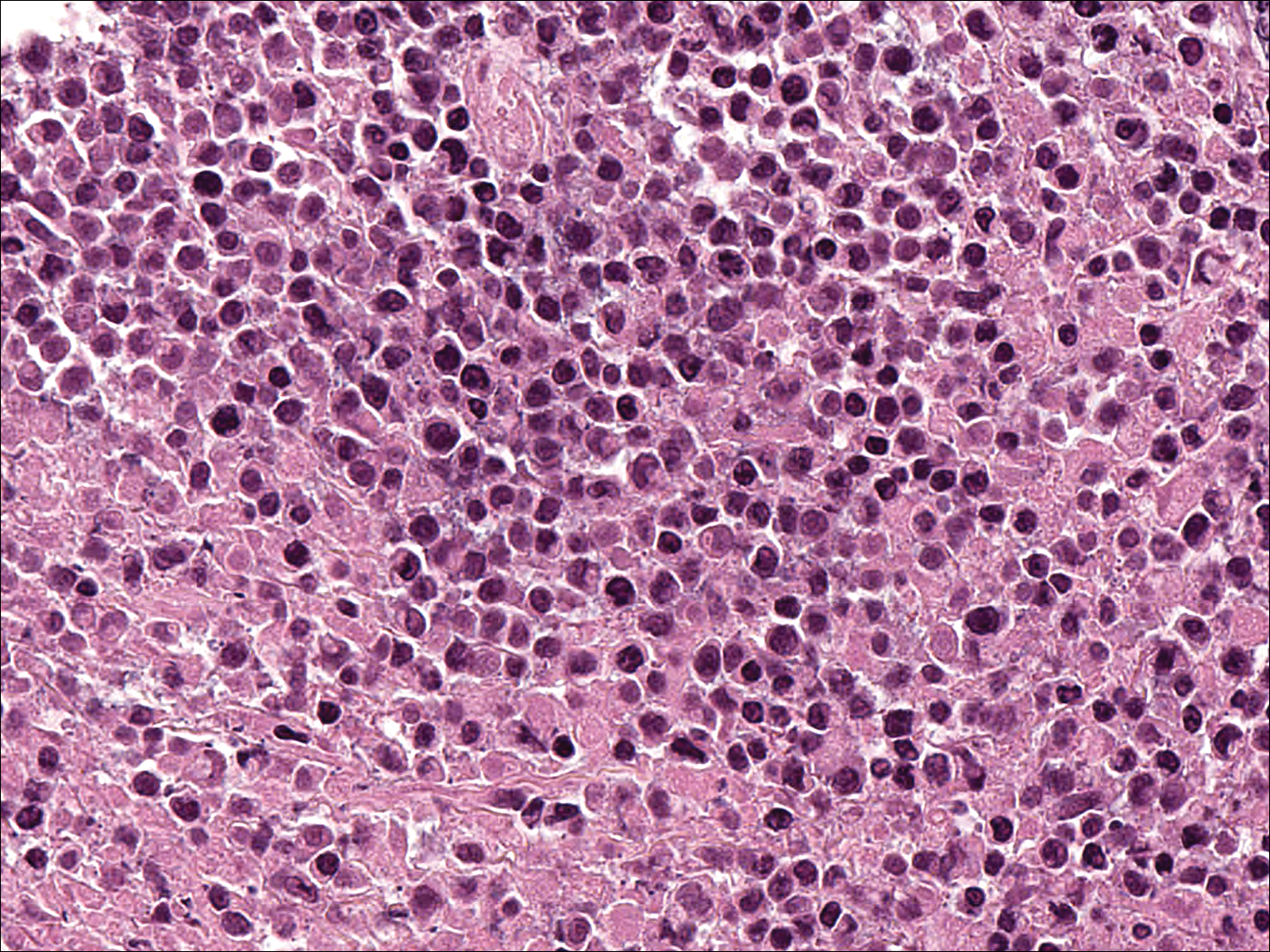
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Practice Points
- Lymphomatoid granulomatosis (LYG) is a rare extranodal angiocentric large B-cell lymphoma driven by the Epstein-Barr virus.
- Lymphomatoid granulomatosis should be suspected when immunocompromised patients present with nodular lung infiltrates and/or nonspecific skin lesions.
- Skin biopsy serves a critical role in establishing the diagnosis of LYG, especially when clinical and radiologic findings are obscured by other comorbidities.