User login
Unsuspected Lymphomatoid Granulomatosis in a Patient With Antisynthetase Syndrome
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
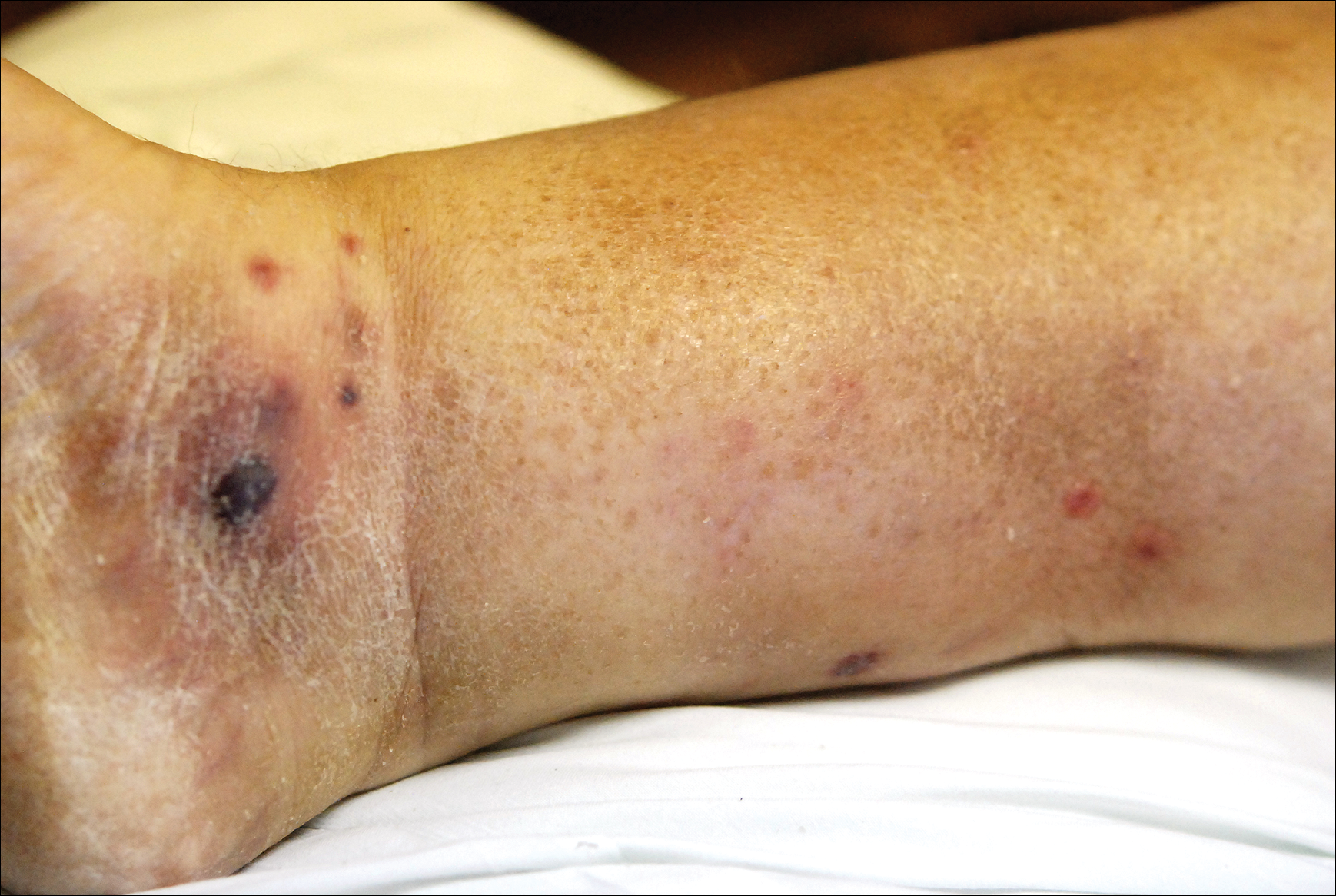
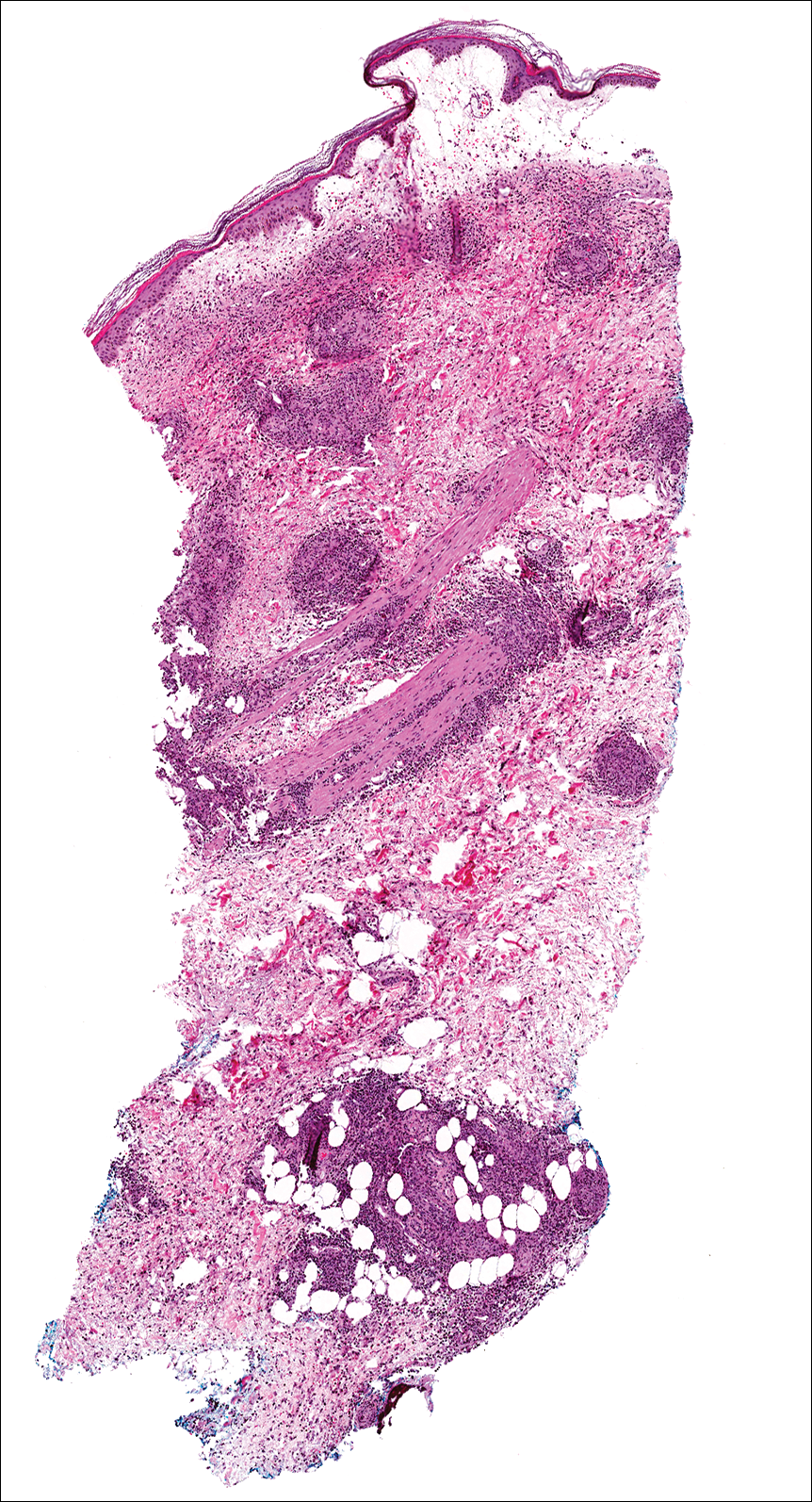
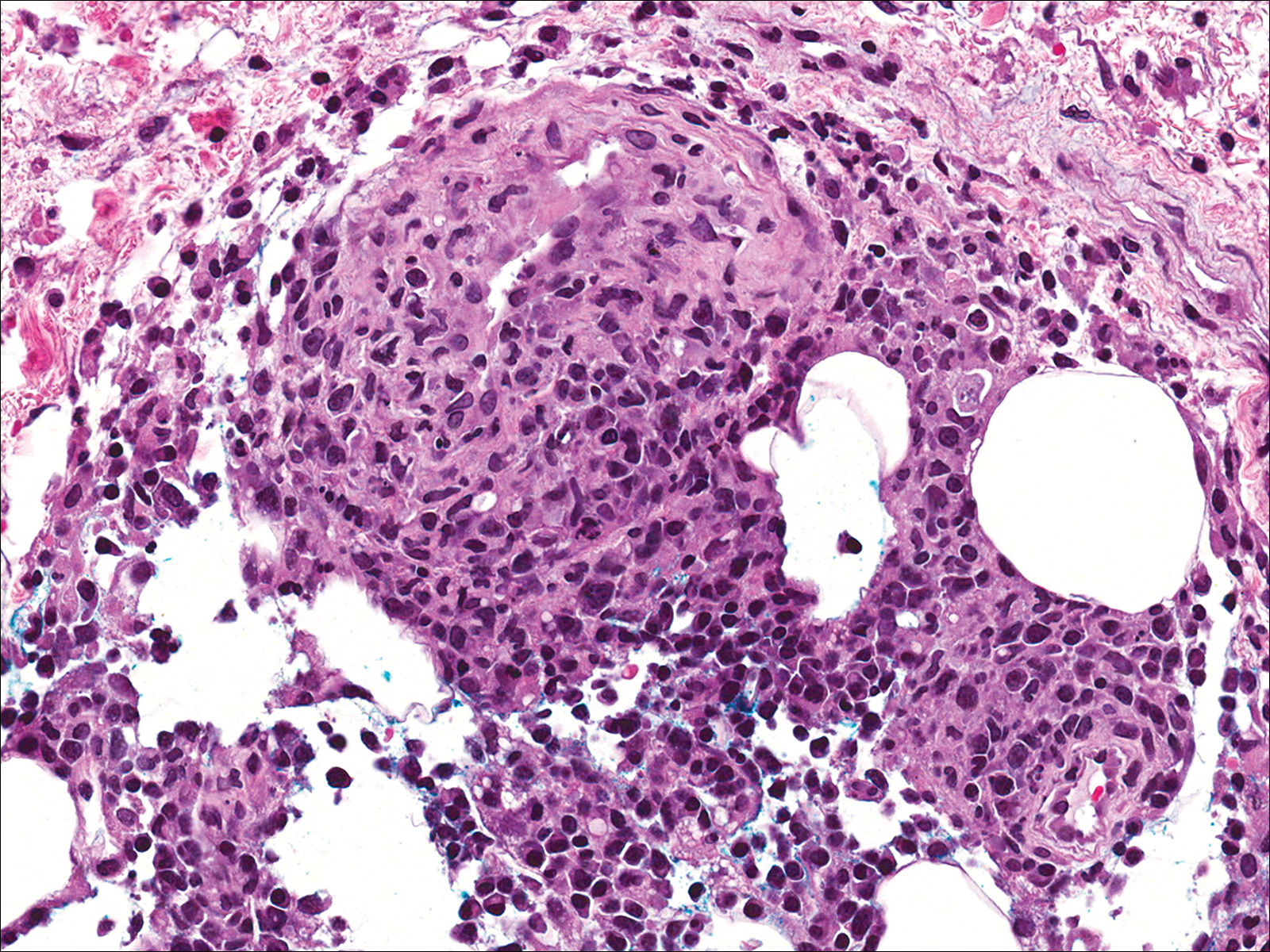
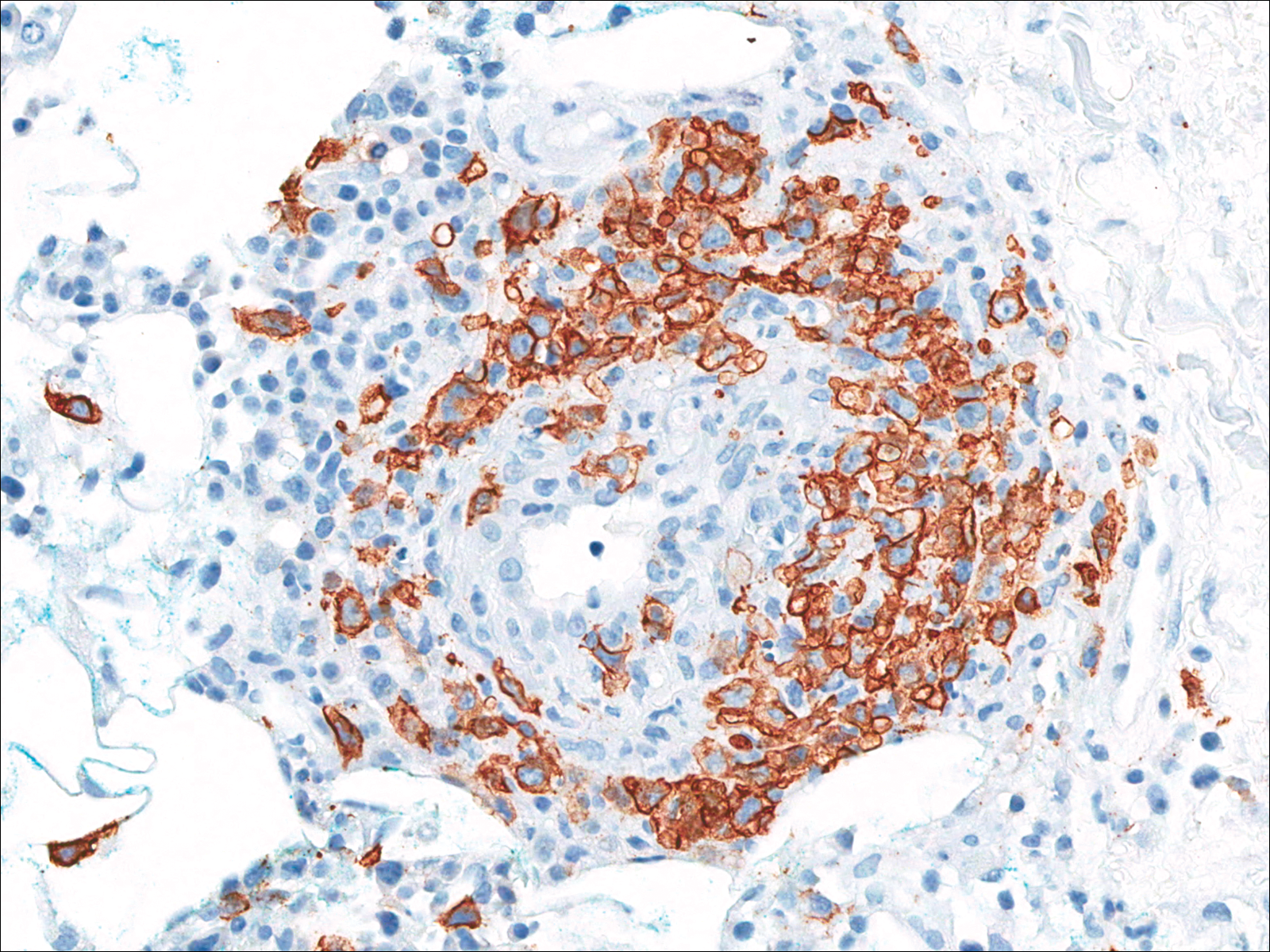
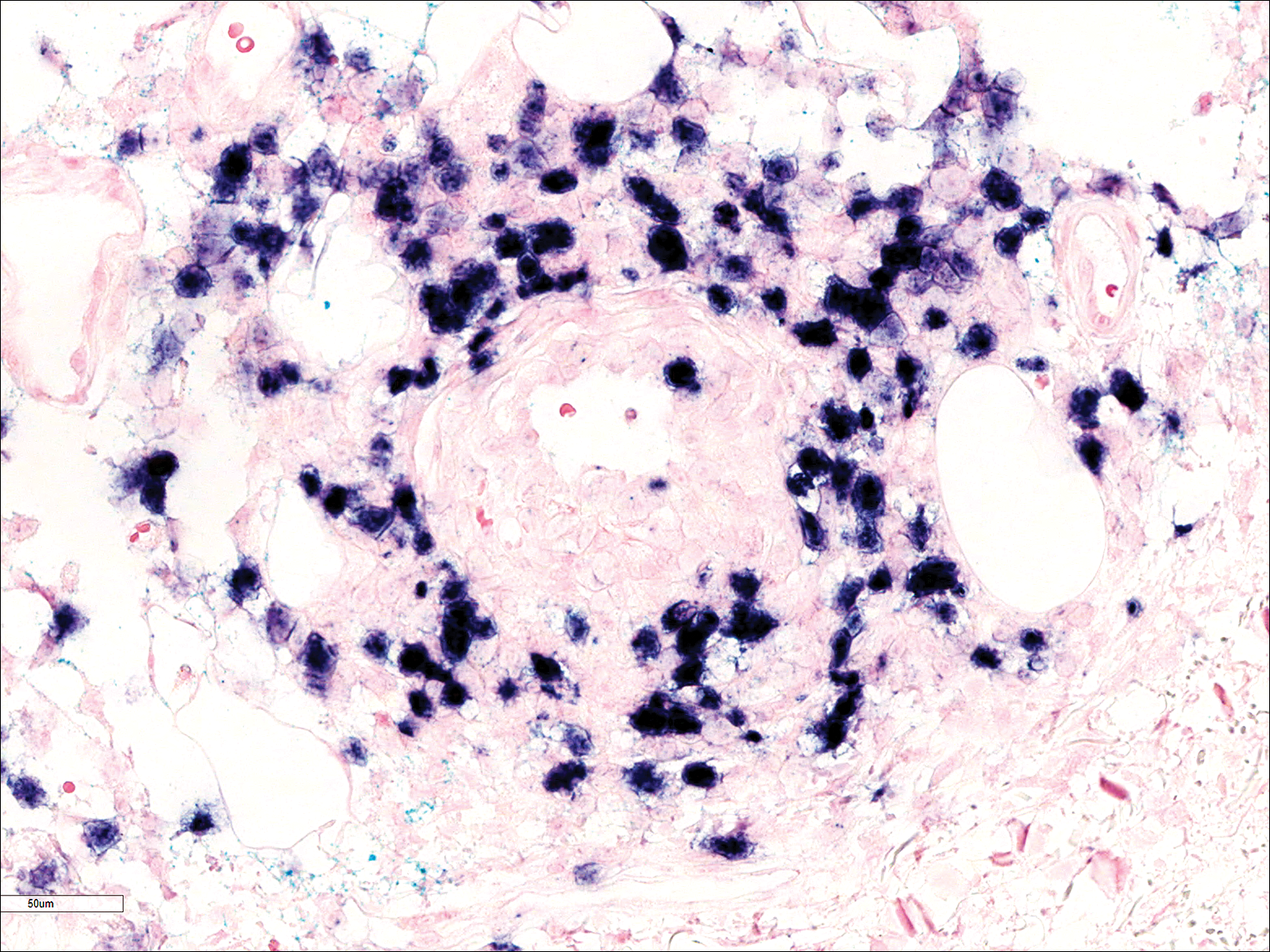
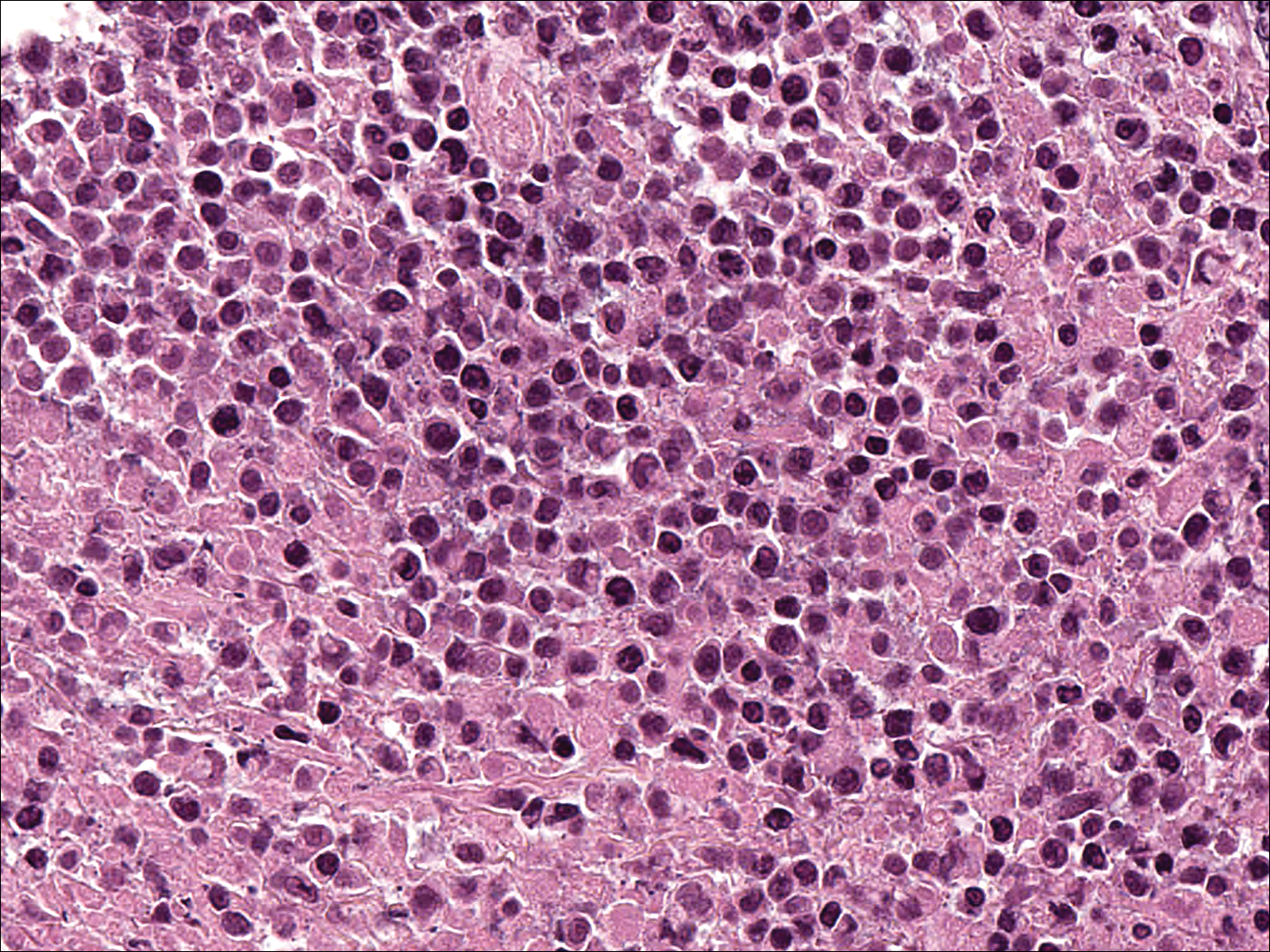
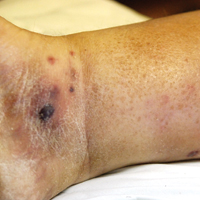
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Practice Points
- Lymphomatoid granulomatosis (LYG) is a rare extranodal angiocentric large B-cell lymphoma driven by the Epstein-Barr virus.
- Lymphomatoid granulomatosis should be suspected when immunocompromised patients present with nodular lung infiltrates and/or nonspecific skin lesions.
- Skin biopsy serves a critical role in establishing the diagnosis of LYG, especially when clinical and radiologic findings are obscured by other comorbidities.
Painful Losses
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
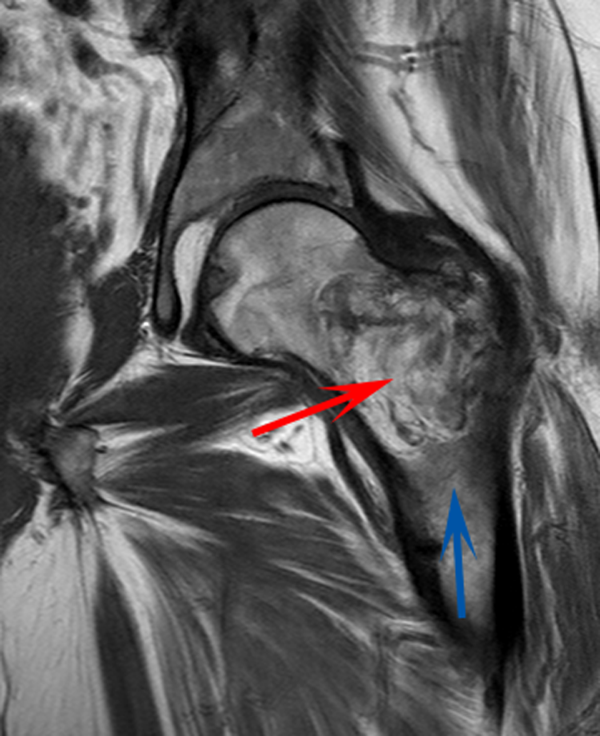
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
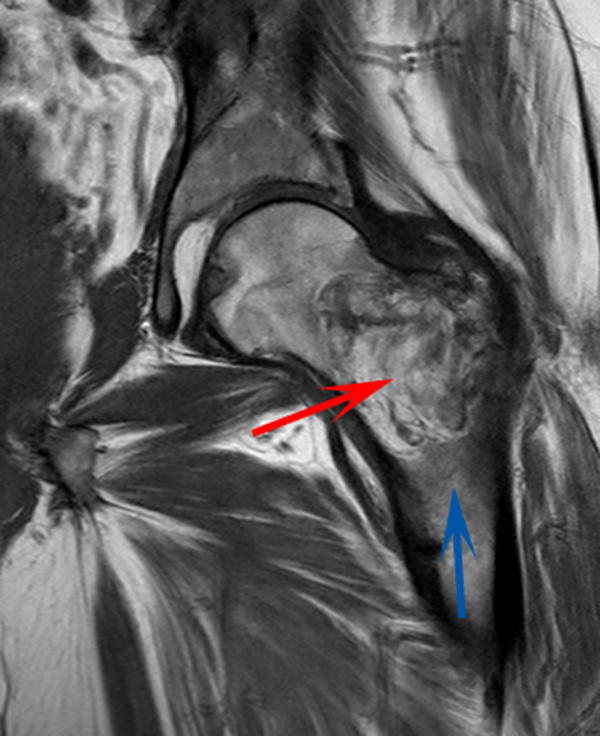
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
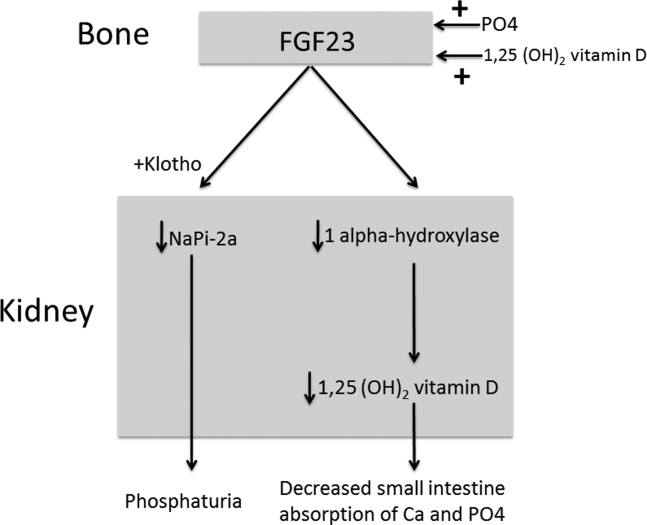
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR < 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.