The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
Author and Disclosure Information
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
Phosphorus binders: The new and the old, and how to choose
Display Headline
Phosphorus binders: The new and the old, and how to choose
Legacy Keywords
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Legacy Keywords
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Serum phosphorus is maintained within normal levels in a tightly regulated system involving interplay between organs, hormones, diet, and other factors.
Dietary phosphorus comes mainly from protein, so restricting phosphorus without introducing protein deficiency is difficult. Food with a low phosphorus-to-protein ratio and plant-based sources of protein may be preferable.
Although dialysis removes phosphorus, it usually does not remove enough, and many patients require phosphorus-binding drugs.
Selection of an appropriate binder should consider serum calcium levels, pill burden, serum iron stores, and cost.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Gate On Date
Wed, 07/25/2018 - 14:45
Un-Gate On Date
Wed, 07/25/2018 - 14:45
Consolidated Pubs: Do Not Show Source Publication Logo
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; nallyj@ccf.org
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; nallyj@ccf.org
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Author and Disclosure Information
Joseph V. Nally, Jr., MD Former Director, Center for Chronic Kidney Disease; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University
Address: Joseph V. Nally, Jr., MD, Glickman Urological and Kidney Institute, Q7, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; nallyj@ccf.org
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
Editor’s note: This Medical Grand Rounds was presented as the 14th Annual Lawrence “Chris” Crain Memorial Lecture, a series that has been dedicated to discussing kidney disease, hypertension, and health care disparities in the African American community. In 1997, Dr. Crain became the first African American chief medical resident at Cleveland Clinic, and was a nephrology fellow in 1998–1999. Dr. Nally was his teacher and mentor.
African Americans have a greater burden of chronic kidney disease than whites. They are more than 3 times as likely as whites to develop end-stage renal disease, even after adjusting for age, disease stage, smoking, medications, and comorbidities. Why this is so has been the focus of much speculation and research.
This article reviews recent advances in the understanding of the progression of chronic kidney disease, with particular scrutiny of the disease in African Americans. Breakthroughs in genetics that help explain the greater disease burden in African Americans are also discussed, as well as implications for organ transplant screening.
ADVANCING UNDERSTANDING OF CHRONIC KIDNEY DISEASE
In the 1990s, dialysis rolls grew by 8% to 10% annually. Unfortunately, many patients first met with a nephrologist on the eve of their first dialysis treatment; there was not yet an adequate way to recognize the disease earlier and slow its progression. And disease definitions were not yet standardized, which led to inadequate metrics and hampered the ability to move disease management forward.
Standardizing definitions
The situation improved in 2002, when the National Kidney Foundation published clinical practice guidelines for chronic kidney disease that included disease definitions and staging.1 Chronic kidney disease was defined as a structural or functional abnormality of the kidney lasting at least 3 months, as manifested by either of the following:
Kidney damage, with or without decreased glomerular filtration rate (GFR), as defined by pathologic abnormalities or markers of kidney damage in the blood, urine, or on imaging tests
Figure 1. Prognosis of chronic kidney disease (CKD) by glomerular filtration rate (GFR) and albuminuria.GFR less than 60 mL/min/1.73 m2, with or without kidney damage.
A subsequent major advance was the recognition that not only GFR but also albuminuria was important for staging of chronic kidney disease (Figure 1).2
Developing large databases
Surveillance and monitoring of chronic kidney disease have generated large databases that enable researchers to detect trends in disease progression.
US Renal Data System. The US Renal Data System has collected and reported on data for more than 20 years from the National Health and Nutrition Examination Survey and the Centers for Medicare and Medicaid Services about chronic and end-stage kidney disease in the United States.
Cleveland Clinic database. Cleveland Clinic has developed a validated chronic kidney disease registry based on its electronic health record.3 The data include demographics (age, sex, ethnic group), comorbidities, medications, and complete laboratory data.4
Alberta Kidney Disease Network. This Canadian research consortium links large laboratory and demographic databases to facilitate defining patient populations, such as those with kidney disease and other comorbidities.
Kaiser Permanente Renal Registry. Kaiser Permanente of Northern California insures more than one-third of adults in the San Francisco Bay Area. The renal registry includes all adults whose kidney function is known. Data on age, sex, and racial or ethnic group are available from the health-plan databases.
DEATHS FROM KIDNEY DISEASE
The mortality rate in patients with end-stage renal disease who are on dialysis has steadily fallen over the past 20 years, from an annual rate of about 25% in 1996 to 17% in 2014, suggesting that care improved during that time. Patients with transplants have a much lower mortality rate: less than 5% annually.5 But these data also highlight the persistent risk faced by patients with chronic kidney disease; even those with transplants have death rates comparable to those of patients with cancer, diabetes, or heart failure.
Death rates correlate with GFR
After the publication of definitions and staging by the National Kidney Foundation in 2002, Go et al6 studied more than 1 million patients with chronic kidney disease from the Kaiser Permanente Renal Registry and found that the rates of cardiovascular events and death from any cause increased with decreasing estimated GFR. These findings were confirmed in a later meta-analysis, which also found that an elevated urinary albumin-to-creatinine ratio (> 1.1 mg/mmol) is an independent predictor of all-cause mortality and cardiovascular mortality.7
Keith et al8 followed nearly 28,000 patients with chronic kidney disease (with an estimated GFR of less than 90 mL/min/1.73 m2) over 5 years. Patients with stage 3 disease (moderate disease, GFR = 30–59 mL/min/1.73 m2) were 20 times more likely to die than to progress to end-stage renal disease (24.3% vs 1.2%). Even those with stage 4 disease (severe disease, GFR = 15–29 mL/min/1.73 m2) were more than twice as likely to die as to progress to dialysis (45.7% vs 19.9%).
Heart disease risk increases with declining kidney function
Navaneethan et al9 examined the leading causes of death between 2005 and 2009 in patients with chronic kidney disease in the Cleveland Clinic database, which included more than 33,000 whites and 5,000 African Americans. During a median follow-up of 2.3 years, 17% of patients died, with the 2 major causes being cardiovascular disease (35%) and cancer (32%) (Table 1). Interestingly, patients with fairly well-preserved kidney function (stage 3A) were more likely to die of cancer than heart disease. As kidney function declined, whether measured by estimated GFR or urine albumin-to-creatinine ratio, the chance of dying of cardiovascular disease increased.
Similar observations were made by Thompson et al10 based on the Alberta Kidney Disease Network database. They tracked cardiovascular causes of death and found that regardless of estimated GFR, cardiovascular deaths were most often attributed to ischemic heart disease (about 55%). Other trends were also apparent: as the GFR fell, the incidence of stroke decreased, and heart failure and valvular heart disease increased.
AFRICAN AMERICANS WITH KIDNEY DISEASE: A DISTINCT GROUP
African Americans constitute about 12% of the US population but account for:
31% of end-stage renal disease
34% of the kidney transplant waiting list
28% of kidney transplants in 2015 (12% of living donor transplants, 35% of deceased donor transplants).
In addition, African Americans with chronic kidney disease tend to be:
Younger and have more advanced kidney disease than whites11
Much more likely than whites to have diabetes, and somewhat more likely to have hypertension
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Figure 2. Risk for all-cause and major cause-specific death in black vs white patients.More likely than whites to die of cardiovascular disease (37.4% vs 34.2%) (Figure 2).9
Overall, the prevalence of chronic kidney disease is slightly higher in African Americans than in whites. Interestingly, African Americans are slightly less likely than whites to have low estimated GFR values (6.2% vs 7.6% incidence of < 60 mL/min/1.73 m2) but are about 50% more likely to have proteinuria (12.3% vs 8.4% incidence of urine albumin-to-creatinine ratio ≥ 30 mg/g).
More likely to be on dialysis, but less likely to die
Although African Americans have only a slightly higher prevalence of chronic kidney disease (about 15% increased prevalence) than whites,12 they are 3 times more likely to be on dialysis.
Nevertheless, for unknown reasons, African American adults on dialysis have about a 26% lower all-cause mortality rate than whites.5 One proposed explanation for this survival advantage has been that the mortality rate in African Americans with chronic kidney disease before entering dialysis is higher than in whites, leading to a “healthier population” on dialysis.13 However, this theory is based on a small study from more than a decade ago and has not been borne out by subsequent investigation.
African Americans with chronic kidney disease: Death rates not increased
African Americans over age 65 with chronic kidney disease have all-cause mortality rates similar to those of whites: about 11% annually. Breaking it down by disease severity, death rates in stage 3 disease are about 10% and jump to more than 15% in higher stages in both African Americans and whites.5
However, African Americans with chronic kidney disease have more heart disease and much more end-stage renal disease than whites.
Disease advances faster despite care
The incidence of end-stage renal disease is consistently more than 3 times higher in African Americans than in whites in the United States.5,14
Multiple investigations have tried to determine why African Americans are disproportionately affected by progression of chronic kidney disease to end-stage renal disease. We recently examined this question in our Cleveland Clinic registry data. Even after adjusting for 17 variables (including demographics, comorbidities, insurance, medications, smoking, and chronic kidney disease stage), African Americans with chronic kidney disease were found to have an increased risk of progressing to end-stage renal disease compared with whites (subhazard ratio 1.38, 95% confidence interval 1.19–1.60).
We examined care measures from the Cleveland Clinic database. In terms of the number of laboratory tests ordered, clinic visits, and nephrology referrals, African Americans had at least as much care as whites, if not more. Similarly, African Americans’ access to renoprotective medicines (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, statins, beta-blockers) was the same as or more than for whites.
Although the frequently attributed reasons surrounding compliance and socioeconomic issues are worthy of examination, they do not appear to completely explain the differences in incidence and outcomes. This dichotomy of a marginally increased prevalence of chronic kidney disease in African Americans with mortality rates similar to those of whites, yet with a 3 times higher incidence of end-stage renal disease in African Americans, suggests a faster progression of the disease in African Americans, which may be genetically based.
GENETIC VARIANTS FOUND
In 2010, two variant alleles of the APOL1 gene on chromosome 22 were found to be associated with nondiabetic kidney disease.15 Three nephropathies are associated with being homozygous for these alleles:
Focal segmental glomerulosclerosis, the leading cause of nephrotic syndrome in African Americans
Hypertension-associated kidney disease with scarring of glomeruli in vessels, the primary cause of end-stage renal disease in African Americans
Human immunodeficiency virus (HIV)- associated nephropathy, usually a focal segmental glomerulosclerosis type of lesion.
The first two conditions are about 3 to 5 times more prevalent in African Americans than in whites, and HIV-associated nephropathy is about 20 to 30 times more common.
African sleeping sickness and chronic kidney disease
Figure 3. Variants in the APOL1 gene that are common in sub-Saharan Africa protect against African sleeping sickness, but homozygosity for these variants increases the risk of chronic kidney disease.The APOL1 variants have been linked to protection from African sleeping sickness caused by Trypanosoma brucei, transmitted by the tsetse fly (Figure 3).16 The pathogen can infect people with normal APOL1 using a serum resistance-associated protein, while the mutant variants prevent or reduce protein binding. Having one variant allele confers protection against trypanosomiasis without leading to kidney disease; having both alleles with the variants protects against sleeping sickness but increases the risk of chronic kidney disease. About 15% of African Americans are homozygous for a variant.17
Retrospective analysis of biologic samples from trials of kidney disease in African Americans has revealed interesting results.
From Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196. Reprinted with permission from Massachusetts Medical Society.
Figure 4. Proportion of patients free from progression of chronic kidney disease, according to APOL1 genotype, in the African American Study of Kidney Disease and Hypertension. The primary outcome was reduction in the glomerular filtration rate (as measured by iothalamate clearance) or incident end-stage renal disease.The African American Study of Kidney Disease and Hypertension (AASK) trial18 evaluated whether tighter blood pressure control would improve outcomes. Biologic samples were available for DNA testing for 693 of the 1,094 trial participants. Of these, 23% of African Americans were found to be homozygous for a high-risk allele, and they had dramatically worse outcomes with greater loss of GFR than those with one or no variant allele (Figure 4). However, the impact of therapy (meeting blood pressure targets, treatment with different medications) did not differ between the groups.
The Chronic Renal Insufficiency Cohort (CRIC) observation study18 enrolled patients with an estimated GFR of 20 to 70 mL/min/1.73 m2, with a preference for African Americans and patients with diabetes. Nearly 3,000 participants had adequate samples for DNA testing. They found that African Americans with the double variant allele had worse outcomes, whether or not they had diabetes, compared with whites and African Americans without the homozygous gene variant.
Mechanism not well understood
The mechanism of renal injury is not well understood. Apolipoprotein L1, the protein coded for by APOL1, is a component of high-density lipoprotein. It is found in a different distribution pattern in people with normal kidneys vs those with nondiabetic kidney disease, especially in the arteries, arterioles, and podocytes.19,20 It can be detected in blood plasma, but levels do not correlate with kidney disease.21 Not all patients with the high-risk variant develop chronic kidney disease; a “second hit” such as infection with HIV may be required.
Investigators have recently developed knockout mouse models of APOL1-associated kidney diseases that are helping to elucidate mechanisms.22,23
EFFECT OF GENOTYPE ON KIDNEY TRANSPLANTS IN AFRICAN AMERICANS
African Americans receive about 30% of kidney transplants in the United States and represent about 15% to 20% of all donors.
Lee et al24 reviewed 119 African American recipients of kidney transplants, about half of whom were homozygous for an APOL1 variant. After 5 years, no differences were found in allograft survival between recipients with 0, 1, or 2 risk alleles.
However, looking at the issue from the other side, Reeves-Daniel et al25 studied the fate of more than 100 kidneys that were transplanted from African American donors, 16% of whom had the high-risk, homozygous genotype. In this case, graft failure was much likelier to occur with the high-risk donor kidneys (hazard ratio 3.84, P = .008). Similar outcomes were shown in a study of 2 centers26 involving 675 transplants from deceased donors, 15% of which involved the high-risk genotype. The hazard ratio for graft failure was found to be 2.26 (P = .001) with high-risk donor kidneys.
These studies, which examined data from about 5 years after transplant, found that kidney failure does not tend to occur immediately in all cases, but gradually over time. Most high-risk kidneys were not lost within the 5 years of the studies.
The fact that the high-risk kidneys do not all fail immediately also suggests that a second hit is required for failure. Culprits postulated include a bacterial or viral infection (eg, BK virus, cytomegalovirus), ischemia or reperfusion injury, drug toxicity, and immune-mediated allograft injury (ie, rejection).
Genetic testing advisable?
Genetic testing for APOL1 risk variants is on the horizon for kidney transplant. But at this point, providing guidance for patients can be tricky. Two case studies27,28 and epidemiologic data suggest that donors homozygous for an APOL1 variant and those with a family history of end-stage kidney disease are at increased risk of chronic kidney disease. Even so, most recipients even of these high-risk organs have good outcomes. If an African American patient needs a kidney and his or her sibling offers one, it is difficult to advise against it when the evidence is weak for immediate risk and when other options may not be readily available. Further investigation is clearly needed into whether APOL1 variants and other biomarkers can predict an organ’s success as a transplant.
The National Institutes of Health are currently funding prospective longitudinal studies with the APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) to determine the impact of APOL1 genetic factors on transplant recipients as well as on living donors. Possible second hits will also be studied, as will other markers of renal dysfunction or disease in donors. Researchers are actively investigating these important questions.
KEEPING SCIENCE RELEVANT
In a recent commentary related to the murine knockout model of APOL1-associated kidney disease, O’Toole et al offered insightful observations regarding the potential clinical impact of these new genetic discoveries.23
As we study the genetics of kidney disease in African American patients, we should keep in mind 3 critical questions of clinical importance:
Will findings identify better treatments for chronic kidney disease? The AASK trial found that knowing the genetics did not affect outcomes of routine therapy. However, basic science investigations are currently underway targeting APOL1 variants which might reduce the increased kidney disease risk among people of African descent.
Should patients be genotyped for APOL1 risk variants? For patients with chronic kidney disease, it does not seem useful at this time. But for renal transplant donors, the answer is probably yes.
How does this discovery help us to understand our patients better? The implications are enormous for combatting the assumptions that rapid chronic kidney disease progression reflects poor patient compliance or other socioeconomic factors. We now understand that genetics, at least in part, drives renal disease outcomes in African American patients.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
References
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28.
Navaneethan SD, Jolly SE, Schold JD, et al. Development and validation of an electronic health record-based chronic kidney disease registry. Clin J Am Soc Nephrol 2011; 6:40–49.
Glickman Urological and Kidney Institute, Cleveland Clinic. 2015 Outcomes. P11.
United States Renal Data System. 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2016.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
Keith D, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Thompson S, James M, Wiebe N, et al; Alberta Kidney Disease Network. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26:2504–2511.
Tarver-Carr ME, Powe NR, Eberhardt MS, et al. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 2002; 13:2363–2370
United States Renal Data System. 2015 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2015; 1:17.
Mailloux LU, Henrich WL. Patient survival and maintenance dialysis. UpToDate 2017.
Burrows NR, Li Y, Williams DE. Racial and ethnic differences in trends of end-stage renal disease: United States, 1995 to 2005. Adv Chronic Kidney Dis 2008; 15:147–152.
Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841–845.
Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-1 efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathogens 2009; 5:e1000685.
Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 2014; 111:E2130–E2139.
Parsa A, Kao WH, Xie D, et al; AASK Study Investigators; CRIC Study Investigators. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183–2196.
Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011; 22:2119–2128.
Hoy WE, Hughson MD, Kopp JB, Mott SA, Bertram JF, Winkler CA. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol 2015; 26:3179–3189.
Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 2014; 25:634–644
Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438.
O’Toole JF, Bruggeman LA, Sedor JR. A new mouse model of APOL1-associated kidney diseases: when traffic gets snarled the podocyte suffers. Am J Kidney Dis 2017; pii: S0272-6386(17)30808-9. doi: 10.1053/j.ajkd.2017.07.002. [Epub ahead of print]
Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 2012; 12:1924–1928.
Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011; 11:1025–1030.
Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15:1615–1622.
Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63:816–819.
Zwang NA, Shetty A, Sustento-Reodica N, et al. APOL1-associated end-stage renal disease in a living kidney transplant donor. Am J Transplant 2016; 16:3568–3572.
Patients with chronic kidney disease are more likely to die than to progress to end-stage disease, and cardiovascular disease and cancer are the leading causes of death.
As kidney function declines, the chance of dying from cardiovascular disease increases.
African Americans tend to develop kidney disease at a younger age than whites and are much more likely to progress to dialysis.
About 15% of African Americans are homozygous for a variant of the APOL1 gene. They are more likely to develop kidney disease and to have worse outcomes.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
Figure 1.
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group(Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD (thomasg3@ccf.org), or Mehdi Shishehbor, DO, MPH (shishem@ccf.org), or visit www.symplifybptrial.com.
References
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA2010; 303:2043–2050.
Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension2011; 57:1076–1080.
Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation2008; 117:e510–e526.
Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc1953; 152:1501–1504.
Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension2009; 54:1195–1201.
Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation1977; 56:691–698.
Kon V. Neural control of renal circulation. Miner Electrolyte Metab1989; 15:33–43.
Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl2000; 75:S2–S6.
Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension1999; 34:724–728.
Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol2004; 287:H695–H703.
Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol1983; 245:F1–F14.
Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet2009; 373:1275–1281.
Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet2010; 376:1903–1909.
Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension2011; 57:911–917.
Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension2011; 58:e19; author replye20.
Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol2011; 58:1176–1182.
Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med2009; 361:932–934.
Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol2011; 100:1095–1101.
Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome?J Hypertens2011; 29:991–996.
Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation2011; 123:1940–1946.
Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol2012; 59:901–909.
Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension2011; 58:559–565.
Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol2012; 101:63–67.
Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol2012; May17[Epub ahead of print]
Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol2011; 100:1049–1057.
Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension2007; 49:839–845.
Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens2003; 16:925–930.
Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens2011; 2011:964394.
Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev2011; 19:52–57.
Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol2012May9. [Epub ahead of print]
George Thomas, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Emmanuel L. Bravo, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Address: George Thomas, MD, Department of Nephrology and Hypertension, Q7, Glickman Urological and Kidney Institute, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail thomasg3@ccf.org
Dr. Shishehbor has disclosed that he has served as a consultant for Medtronic.
George Thomas, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Emmanuel L. Bravo, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Address: George Thomas, MD, Department of Nephrology and Hypertension, Q7, Glickman Urological and Kidney Institute, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail thomasg3@ccf.org
Dr. Shishehbor has disclosed that he has served as a consultant for Medtronic.
Author and Disclosure Information
George Thomas, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Heart and Vascular Institute, Cleveland Clinic
Emmanuel L. Bravo, MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Glickman Urological and Kidney Institute, Cleveland Clinic
Address: George Thomas, MD, Department of Nephrology and Hypertension, Q7, Glickman Urological and Kidney Institute, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail thomasg3@ccf.org
Dr. Shishehbor has disclosed that he has served as a consultant for Medtronic.
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
Figure 1.
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group(Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD (thomasg3@ccf.org), or Mehdi Shishehbor, DO, MPH (shishem@ccf.org), or visit www.symplifybptrial.com.
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
Figure 1.
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group(Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD (thomasg3@ccf.org), or Mehdi Shishehbor, DO, MPH (shishem@ccf.org), or visit www.symplifybptrial.com.
References
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA2010; 303:2043–2050.
Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension2011; 57:1076–1080.
Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation2008; 117:e510–e526.
Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc1953; 152:1501–1504.
Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension2009; 54:1195–1201.
Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation1977; 56:691–698.
Kon V. Neural control of renal circulation. Miner Electrolyte Metab1989; 15:33–43.
Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl2000; 75:S2–S6.
Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension1999; 34:724–728.
Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol2004; 287:H695–H703.
Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol1983; 245:F1–F14.
Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet2009; 373:1275–1281.
Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet2010; 376:1903–1909.
Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension2011; 57:911–917.
Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension2011; 58:e19; author replye20.
Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol2011; 58:1176–1182.
Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med2009; 361:932–934.
Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol2011; 100:1095–1101.
Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome?J Hypertens2011; 29:991–996.
Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation2011; 123:1940–1946.
Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol2012; 59:901–909.
Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension2011; 58:559–565.
Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol2012; 101:63–67.
Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol2012; May17[Epub ahead of print]
Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol2011; 100:1049–1057.
Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension2007; 49:839–845.
Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens2003; 16:925–930.
Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens2011; 2011:964394.
Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev2011; 19:52–57.
Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol2012May9. [Epub ahead of print]
References
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA2010; 303:2043–2050.
Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension2011; 57:1076–1080.
Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation2008; 117:e510–e526.
Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc1953; 152:1501–1504.
Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension2009; 54:1195–1201.
Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation1977; 56:691–698.
Kon V. Neural control of renal circulation. Miner Electrolyte Metab1989; 15:33–43.
Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl2000; 75:S2–S6.
Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension1999; 34:724–728.
Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol2004; 287:H695–H703.
Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol1983; 245:F1–F14.
Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet2009; 373:1275–1281.
Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet2010; 376:1903–1909.
Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension2011; 57:911–917.
Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension2011; 58:e19; author replye20.
Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol2011; 58:1176–1182.
Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med2009; 361:932–934.
Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol2011; 100:1095–1101.
Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome?J Hypertens2011; 29:991–996.
Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation2011; 123:1940–1946.
Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol2012; 59:901–909.
Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension2011; 58:559–565.
Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol2012; 101:63–67.
Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol2012; May17[Epub ahead of print]
Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol2011; 100:1049–1057.
Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension2007; 49:839–845.
Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens2003; 16:925–930.
Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens2011; 2011:964394.
Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev2011; 19:52–57.
Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol2012May9. [Epub ahead of print]
Renal sympathetic nerves help regulate volume and blood pressure as they innervate the renal tubules, blood vessels, and juxtaglomerular apparatus. They carry both afferent and efferent signals between the central nervous system and the kidneys.
Surgical sympathectomy was done in the 1950s for malignant hypertension. It had lasting antihypertensive results but also caused severe procedure-related morbidity. A new percutaneous procedure for selective renal denervation offers the advantage of causing few major procedure-related adverse effects.
Selective renal denervation decreases norepinephrine spillover and muscle sympathetic nerve activity, evidence that the procedure reduces sympathetic tone.
The major clinical trials done so far have found that renal denervation lowers blood pressure significantly, and the reduction is sustained for at least 3 years.
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, The Cleveland Clinic
Address: Joseph V. Nally, Jr., MD, Department of Nephrology and Hypertension, A51, The Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195
Medical Grand Rounds articles are based on edited transcripts from Division of Medicine Grand Rounds presentations at The Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, The Cleveland Clinic
Address: Joseph V. Nally, Jr., MD, Department of Nephrology and Hypertension, A51, The Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195
Medical Grand Rounds articles are based on edited transcripts from Division of Medicine Grand Rounds presentations at The Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Author and Disclosure Information
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, The Cleveland Clinic
Address: Joseph V. Nally, Jr., MD, Department of Nephrology and Hypertension, A51, The Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195
Medical Grand Rounds articles are based on edited transcripts from Division of Medicine Grand Rounds presentations at The Cleveland Clinic. They are approved by the author but are not peer-reviewed.






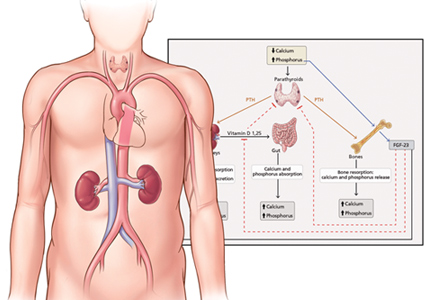



 Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.
Adapted from Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Nally JV Jr. Cause-specific deaths in non-dialysis-dependent CKD. J Am Soc Nephrol 2015; 26:2512–2520.