User login
Importance of platelets and platelet response in acute coronary syndromes
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
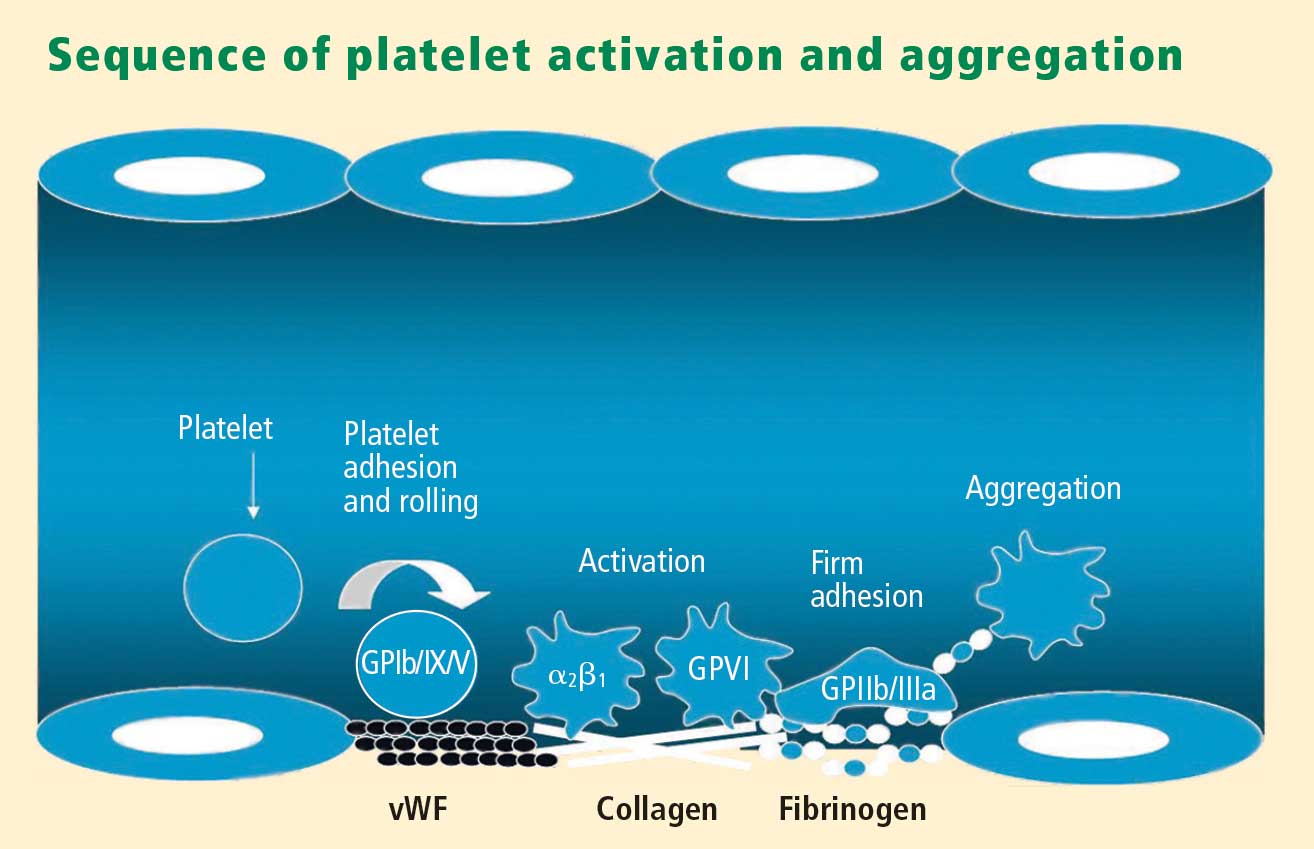
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
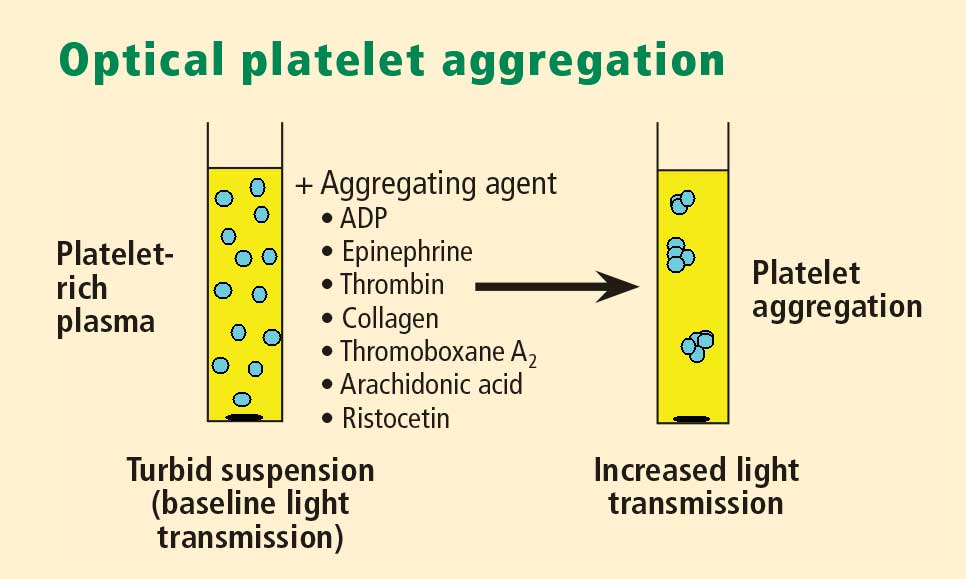
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
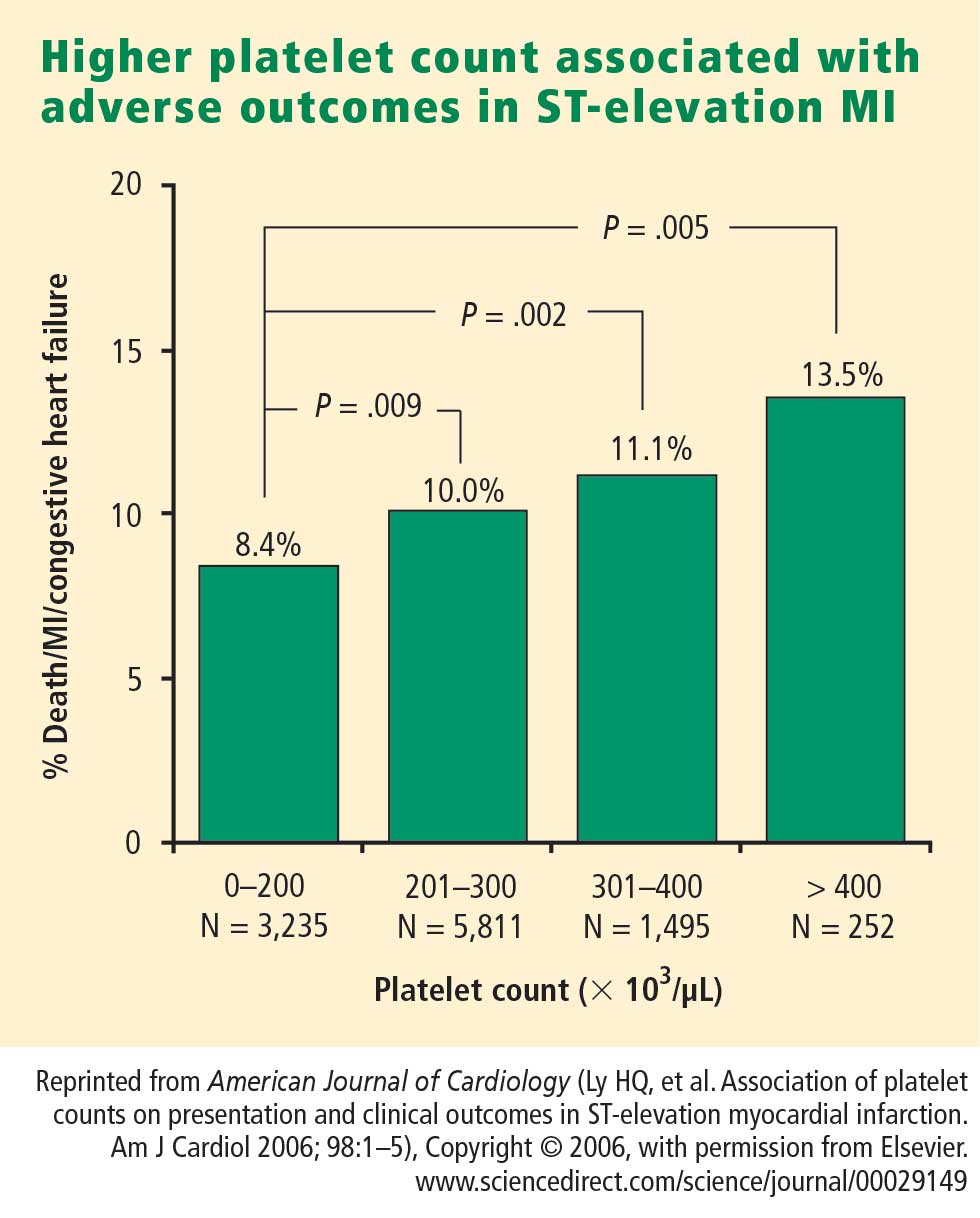
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
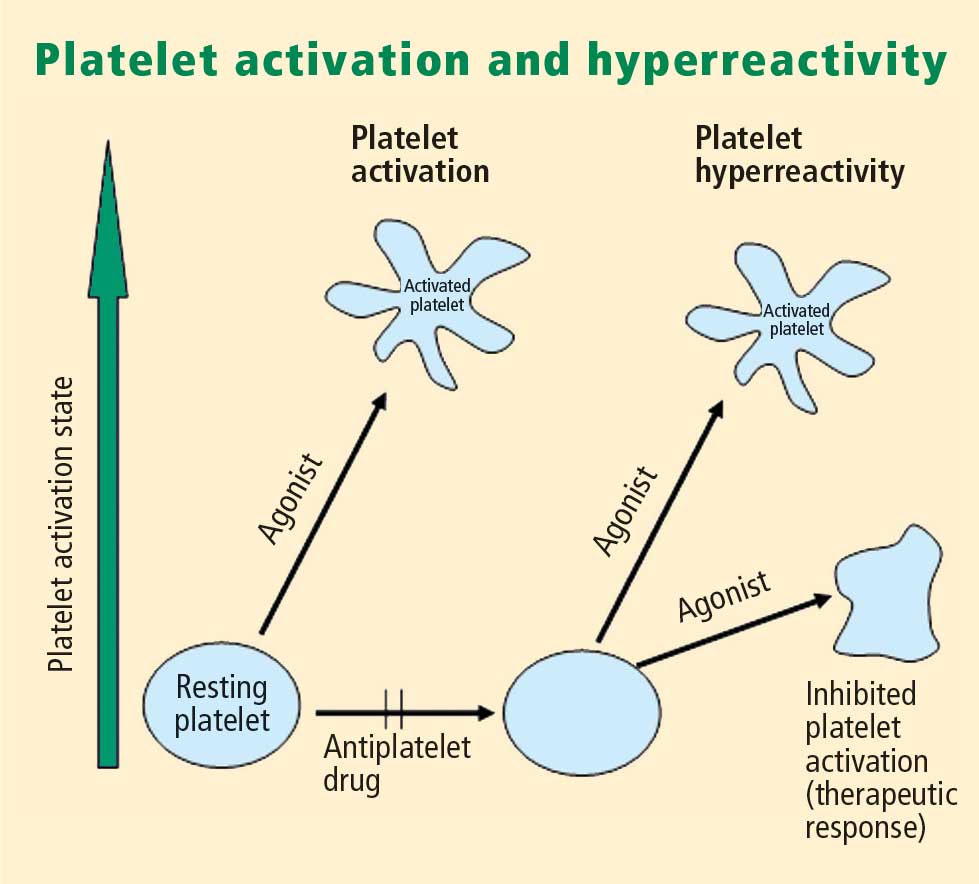
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
KEY POINTS
- Platelet function assays are inherently variable because they measure cell function rather than a single analyte.
- Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of different aspects of platelet function.
- There appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major cardiac events associated with platelet hyperreactivity.
- For predicting cardiac events, receiver operating characteristic (ROC) curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrary cutoff values.
Platelet response in practice: Applying new insights and tools for testing and treatment
CASE STUDY: THROMBOSIS AFTER STENTING DESPITE ANTIPLATELET THERAPY
Dr. Deepak Bhatt: We have taken in a wealth of terrific information from the three preceding talks in this symposium. Let’s now share some questions from the audience and explore some of the points raised in the preceding talks in a bit more practical detail for clinicians. Our three prior speakers are joined in this panel discussion by Cleveland Clinic’s Dr. Frank Peacock, who brings an emergency medicine perspective.
Let’s begin with a case-based question supplied from the audience. The patient is a 42-year-old morbidly obese man without diabetes who had a non-ST-elevation myocardial infarction (MI) less than 1 year ago. A drug-eluting stent was placed at the time of his MI, and now restenosis has occurred. He is on aspirin and clopidogrel 75 mg/day. Do you recommend running a vasodilator-stimulated phosphoprotein (VASP) test and possibly increasing the clopidogrel dose to 150 mg/ day, or should the patient just be switched to prasugrel (assuming it is commercially available) without running the VASP test?
I’ll take a quick initial stab at this question. Studies of antiplatelet therapies to prevent instent restenosis have been a mixed bag. Some of the trials with glycoprotein IIb/IIIa inhibitors have shown an effect on restenosis, but most have not. Similarly, some of the analyses of the thienopyridines ticlopodine and clopidogrel have shown an effect on restenosis, but most have not.
For the most part, restenosis does not appear to be heavily mediated by platelets, at least not in a way that we can influence by therapy. On the other hand, stent thrombosis is highly platelet mediated, so I would alter the case to one in which stent thrombosis is the clinical problem. Assuming that the patient has been adherent to his antiplatelet regimen, which tests would you perform, and how would you act on the information from those tests?
Dr. Kandice Kottke-Marchant: The 2007 guidelines on acute coronary syndrome (ACS) management from the American College of Cardiology and American Heart Association (ACC/AHA)1 do not address platelet function testing, and almost none of the clinical trials of antiplatelet agents had an arm that included testing and dose adjustment based on platelet function studies. Platelet testing is available at some centers; at Cleveland Clinic, we use platelet aggregation testing. One can do platelet aggregation testing on a patient-by-patient basis; if inhibition appears to be suboptimal, a treatment decision should be made, but there is little guidance from the literature to steer that decision. I have seen clinicians increase the dose of clopidogrel or aspirin in response to platelet function tests, which occasionally triggers a confirmatory call from the pharmacy department.
Dr. Bhatt: When I was still at Cleveland Clinic, our chief medical resident did an analysis of platelet function testing, and it was remarkable how much testing was performed and how often it changed management, largely in the absence of any outcomes data, as Dr. Kottke-Marchant pointed out. Dr. Alexander, what are your recommendations with respect to platelet function testing today?
Dr. John Alexander: The case you describe is one in which applying evidence is not easy. There are no trials to supply any evidence to change therapy in this patient, a morbidly obese man receiving 75 mg/day of clopidogrel. There is certainly a rationale, however, to believe that a standard “one size fits all” 75-mg daily dose of clopidogrel may not be enough for him. The trade-off with a higher dosage is a higher risk of bleeding, however, so I would first be sure that he has been adherent to his current regimen of clopidogrel and aspirin.
Dr. Bhatt: Is there a role for point-of-care testing to determine whether he is adherent to the medicines?
Dr. Kottke-Marchant: Several of the point-of-care tests, such as the VerifyNow rapid platelet function analyzer, have specific cartridges for aspirin and for clopidogrel. If platelets were not being inhibited, it would suggest that the doses were too low, given the patient’s weight, but you probably would not be able to determine whether he was resistant to clopidogrel.
Dr. W. Frank Peacock: One way to verify that patients are taking their aspirin is to take a small urine sample and squirt in 2 mL of ferric chloride. If the sample turns purple, it means they are taking their aspirin. Once that is established, you can try to determine whether the drug is working on their platelets.
Dr. Alexander: Another potential explanation for stent thrombosis is faulty stent placement. In this case I would consider asking an interventional colleague to perform intravascular ultrasonography to make sure the stent was implanted properly before I changed the patient’s antithrombotic therapy.
Dr. Bhatt: That’s a great technical point. We always want to make sure that a case of stent thrombosis is not due to a mechanical problem. We should be asking: Is the stent properly sized and well opposed? Is there a distal dissection or any other issue that could predispose to stent thrombosis?
Dr. Alexander: This case illustrates a host of other challenges that underscore how much more work we need to do to define optimal antiplatelet therapy. Suppose we perform platelet function testing and find a low level of platelet inhibition in this patient with stent thrombosis, and we change his antiplatelet regimen. When should we test him again? If we retest in 3 months and find that he has a higher than expected level of platelet inhibition on the new antiplatelet regimen, do we dial down the intensity? Once again, there is no evidence to guide these decisions, and levels of platelet inhibition are driven not just by the medications but also by what is going on in the patient’s platelets—it is quite multifactorial.
POINT-OF-CARE PLATELET FUNCTION TESTING: CURRENT LIMITS, FUTURE ROLES
Dr. Bhatt: While we’re discussing platelet function testing, I found it interesting, Dr. Kottke-Marchant, that you said the use of bleeding time as a platelet test is finally going away. Testing of bleeding time has been around forever, but I agree that it doesn’t have much value in clinical practice. Do you think bleeding time will continue to have any role in drug development? Most phase 2 trials, and certainly phase 1 trials, still capture bleeding time to assess whether or not a drug is working. Should that, too, be jettisoned, or does bleeding time still have some merit in this context?
Dr. Kottke-Marchant: I would jettison it in drug development as well because of the considerable variability in bleeding time. It is not a test that can be standardized, and no quality control can be done. The results depend on skin turgor, age, and many other variables.
We need a global assay that will pick up multiple aspects of platelet function, such as flow-based adhesion, aggregation, and granule release. The bleeding time is a shear-dependent test, whereas the platelet aggregation test that is used in most drug trials is an artificial assay that measures only aggregation, but not under shear. The VerifyNow rapid platelet function analyzer does not measure platelets under shear and is not a global assay.
Dr. Marc Sabatine: I would underscore the need for a reliable point-of-care test of platelet function. When we prescribe a statin or an antihypertensive drug, we don’t just send the patient out the door and hope that everything will be okay. We measure the response, knowing that genotype, environmental factors, or medication factors can affect the response. When we prescribe an antiplatelet drug, we need a reliable point-of-care device to make certain that the patient is getting appropriate platelet inhibition.
I am reminded of a recent study of point-of-care measurement of platelet inhibition in patients receiving clopidogrel prior to nonemergent percutaneous coronary intervention (PCI).2 Rather than just treating patients with PCI and sending them out the door, the investigators kept giving patients clopidogrel and measuring their platelet inhibition until they achieved an appropriate degree of inhibition, after which PCI was performed. Event rates were significantly reduced in the patient group treated this way, which suggests a need to individualize therapy and move away from the “one size fits all” mindset.
Dr. Bhatt: Dr. Peacock, you’ve led a study of point-of-care assays in the emergency department. What might ultimately be the role of point-of-care testing in emergency medicine, and might it influence drug selection?
Dr. Peacock: My short answer is that I think there will be a role for point-of-care testing, with all the caveats that have been discussed. There may even be a day when we do genetic testing and look for DNA. Honestly, though, I’m somewhat of a skeptic because I’m not looking at the genetics. I see many patients who do crack cocaine who come to the emergency room with chest pain and have risk factors, but I send these patients home because they are not having an event. The real question is, “Is it an event?” If a patient is having an event and he or she has platelet resistance or hyperreactivity—whatever we term it—then you have to decide the next step.
As you mentioned, we just completed a study that evaluated a couple hundred patients for platelet inhibition resistance to aspirin, and one finding was that the incidence of platelet resistance to aspirin was much lower than we had anticipated. Studies from the literature suggest that the prevalence of resistance is around 30%, but in our study it was 6.5%.3
Dr. Kottke-Marchant: It depends on how and in whom you measure resistance. Different tests will give you different numbers. Even among studies using the same measurement techniques, the results depend on the patient population. If it’s a fairly stable cardiac population, you may see aspirin resistance rates of 4% or 5%. If it’s a population of patients who have had multiple MIs, the rate may be higher.
Dr. Peacock: That’s exactly my point. In the emergency department we see a mixed bag. We see many people who have had no prior events and have no acute event occurring. So in that setting you are going to get results that suggest that no intervention is required, whereas in that small percentage of patients in whom something is happening, your drug choice may be different.
Dr. Alexander: We are still talking about resistance to antiplatelet drugs as though it were a patient-level variable, but it’s my impression that it changes over time and within a patient.
Dr. Kottke-Marchant: It can change over time. There aren’t many good longitudinal studies. Most of the studies of “aspirin resistance” are really snapshot studies with measurements taken at one point in time. A term I prefer is “platelet reactivity.” To really assess treatment efficacy, we are going to have to look at the basal level of platelet reactivity.
WHAT ROLE FOR GENOTYPING IN GUIDING ANTIPLATELET THERAPY?
Dr. Bhatt: Dr. Peacock alluded to a potential role for genetic testing. Dr. Sabatine, you have done a lot of interesting work with genotyping in the TRITON-TIMI 38 study of prasugrel and clopidogrel. What is the future role of genotyping in determining which antiplatelet therapy is best for which patient?
Dr. Sabatine: As I mentioned, cytochrome P450 enzymes play a critical role in the metabolism of clopidogrel. These enzymes are fairly polymorphic—mutations in their encoding genes are responsible for subtle changes in effect, unlike the traditional mutations that we think about for sickle cell disease, for example. A wealth of data has been published showing that genetic variants are associated with decreased functional activity of cytochrome P450 enzymes, demonstrating the pharmacologic importance of these variants.
Individuals who carry variant alleles appear to respond differently to clopidogrel. A variety of small studies show that those who carry specific variants—particularly in the CYP2C19 enzyme, but in other enzymes as well—appear to have a diminished response to clopidogrel. There are also data showing that individuals with a diminished response to clopidogrel have worse outcomes.4 Our group is studying the impact of genetic variants that decrease the functional activity of cytochrome P450 enzymes on clinical outcomes. (Editor’s note: This study has since been published by Mega et al.5)
The practical implication may lie in point-of-care genotyping, which appears possible and will be clinically useful if a strong link can be demonstrated between genotype and outcomes. If point-of-care genotyping becomes practical, it will raise the question of whether both genotyping and platelet aggregation testing are needed. I think they might indeed be complementary in risk prediction, as is the case with genetic variants that affect low-density lipoprotein cholesterol (LDL-C) levels. In the lipid arena, we have seen that genetic effects and lipid levels provide independent incremental information about risk. That’s because of the high degree of variation in LDL-C levels: an LDL-C measurement is a snapshot in time, yet a variety of factors can influence LDL-C levels. In contrast, genotype is an invariant factor. Similarly, in the platelet arena, platelet aggregation studies and genotyping may be synergistic in predicting an individual’s predisposition to events and response to medications.
Dr. Bhatt: While we’re discussing pathways of metabolism, the literature, though scant, suggests a potential interaction between proton pump inhibitors and clopidogrel. I was co-chair of a recent American College of Cardiology/ American Heart Association/American College of Gastro-enterology consensus document that endorsed liberal use of proton pump inhibitors in patients who are at gastrointestinal risk, including those on antiplatelet therapy.6 The gastroenterologists believed strongly that proton pump inhibitors were safe and in fact underused in these patients. What do you think about the clopidogrel–proton pump inhibitor interaction? Should we be concerned?
Dr. Sabatine: Proton pump inhibitors are not only substrates for, but also inhibitors of, CYP2C19, a key enzyme that helps transform clopidogrel into an active metabolite. For this reason, there has been interest in whether concomitant use of proton pump inhibitors would blunt the efficacy of clopidogrel. The same concern was raised about giving clopidogrel with certain statin drugs that are also metabolized by the cytochrome P450 system, and several studies have shown an effect of these statins on clopidogrel’s platelet inhibition. However, there is no evidence that coadministration of these statins has affected clinical outcomes with clopidogrel in clinical trials. So it may be that while competition for the cytochrome P450 system is one factor, it’s not enough of a factor to tip the scale and result in a clinical event. The same may be true of coadministration of proton pump inhibitors; meanwhile, we await definitive data that concomitant use with clopidogrel leads to higher rates of ischemic events.
DIAGNOSTIC UNCERTAINTY IN THE EMERGENCY SETTING
Dr. Bhatt: We heard about quite a few new antiplatelet drugs in Dr. Sabatine’s presentation, some of which will likely be taken up in clinical practice. Dr. Peacock, from an emergency department perspective, how will you integrate all these new agents with the numerous therapies already available? What should emergency departments do to come to grips with and ultimately take advantage of these different forms of therapy as well as emerging platelet function tests?
Dr. Peacock: The piece that’s unique or especially pertinent to the emergency department is diagnostic uncertainty. Diagnosis and management are easy when a patient has an ST-elevation MI because we all know what that looks like and we know what to do in response. To some extent non-ST-elevation MI is fairly simple too. ACS is a lot more difficult because we don’t have a good definition for unstable angina, and that’s where diagnosis and management become problematic. And with high-sensitivity troponins coming out now, the question of non-ST-elevation MI is going to get more and more confusing because we will have a lot more patients who meet criteria without having an acute coronary artery event.
So it is going to be important that studies be designed correctly. A lot of the studies reviewed today were efficacy studies, showing that a particular drug works well in a carefully defined population, but they were not efficiency studies: they did not take into account the real-world diagnostic uncertainty—and inevitable misdiagnoses—that emergency departments encounter before starting therapy.
Take the CURE trial, for example. It was a great study, showing that clopidogrel reduced the hazard ratio for major coronary events by 20% in patients with unstable angina,7 and the message was that everybody should be using clopidogrel. A close look at the study, however, reveals that about half the patients did not receive clopidogrel in the emergency department. When patients did receive it early, it was driven by the cardiologist, who was absolutely certain of the diagnosis. But if the study was not designed to test early use, then it is a big leap to extrapolate its findings to this circumstance.
Many of the patients in the CURE trial were enrolled the day after presentation, when their diagnosis was certain—ie, they had a rise in troponin after their symptoms. But when a patient first arrives in the emergency department, we are not certain of the diagnosis. And if we use a drug such as clopidogrel, with a duration of action as long as 5 days, we have committed the entire medical system to a certain course of management for that period of time. If we get the diagnosis wrong, this commitment could restrict management options for up to 5 days.
The question for emergency physicians becomes, “How long is long enough to know whether I can pull the trigger on a therapy and be correct?” With all the new drugs coming along, the way to answer this is to do efficiency studies in a real-world environment in addition to efficacy studies.
Dr. Alexander: Yes, one of the biggest limitations of antiplatelet drug studies to date is that they usually haven’t really addressed the timing of drug initiation. We often assume that if a drug is shown to be beneficial, then it should be started as soon as possible. As we just heard, that may have been an unfounded extrapolation from the CURE trial. The same sort of thing happened with the ISIS trial of aspirin in patients with ST-elevation MI.8 In response to the ISIS results, clinicians rushed to give patients aspirin right away even though many of the patients in the trial may have received their aspirin the day after presentation. For these reasons, the EARLY-ACS study,9 which is addressing a very simple question—whether early upstream use of glycoprotein IIb/IIIa inhibitors is beneficial—has been a challenging trial to complete.
WHAT ROLE FOR THIENOPYRIDINE PRETREATMENT?
Dr. Bhatt: Dr. Sabatine, you presented data from the large TRITON-TIMI 38 trial comparing prasugrel with clopidogrel. I’m interested in how you would use prasugrel in practice, assuming it receives marketing approval, especially in light of its bleeding risk, particularly in patients in whom coronary artery bypass graft surgery (CABG) is planned. Many hospitals pretreat patients with clopidogrel in the emergency department. How would you manage a patient who shows up in the emergency room with ACS? Would you give clopidogrel, would you wait and give prasugrel, or would you do something else? If you gave clopidogrel, what loading dose would you use—300 mg, 600 mg, or, as some have suggested, 900 or 1,200 mg?
Dr. Sabatine: I am a strong proponent of pretreatment. Data from multiple studies show a benefit to this strategy, and even the original CURE trial showed a roughly 30% reduction in ischemic events within the first 24 hours of clopidogrel initiation.7
I think the dosing strategy depends on how the patient is going to be managed. If management is going to be conservative, then I would start the patient on 300 mg of clopidogrel when he or she came in. If the patient is going to the cardiac catheterization laboratory in a few hours, I would pretreat with 600 mg of clopidogrel. For prasugrel, the need for pretreatment is less clear, given the drug’s faster onset of action and greater degree of platelet inhibition. In the TRITON-TIMI 38 study,10 prasugrel was given, by and large, after diagnostic angiography, and thus one could use that approach in practice.
In terms of clopidogrel versus prasugrel, I would embrace prasugrel for the large majority of my patients, being mindful of the risk of bleeding. I would not hesitate to give the medication to diabetics or to younger, more robust patients. The 50% reduction in stent thrombosis with prasugrel versus clopidogrel in TRITON-TIMI 38 is huge,11 given that the risk of death with stent thrombosis is probably 25% or greater. So I would want to have prasugrel on board to reduce the risk of stent thrombosis, especially if a drug-eluting stent were being implanted.
Dr. Bhatt: Dr. Alexander, let’s get your take on a similar scenario. Assuming that prasugrel gains marketing approval, how would you manage patients with non-ST-elevation MI who present to the emergency department? Would you pretreat with clopidogrel? Would you wait until angiography and then, depending on the anatomy, treat with prasugrel? Or would you potentially pretreat with prasugrel, which has not been studied and would not be a labeled indication? How would you reconcile the data?
Dr. Alexander: At Duke, I expect that prasugrel will not be used prior to the catheterization laboratory in patients with non-ST-elevation ACS due to concerns about whether the patients will undergo PCI or be managed medically or with CABG.
Dr. Bhatt: That makes sense, since there was a fair amount of bleeding with prasugrel in those patients in TRITON-TIMI 38.
Dr. Alexander: Correct. Moreover, at Duke we don’t use as much upstream clopidogrel as we would, based on the evidence, if I were managing all the patients. There is still a lot of pushback about upstream clopidogrel from our surgeons because patients are going to surgery quickly these days, sometimes just a day after catheterization, and that’s when a loading dose of clopidogrel can be problematic. We are also still fairly heavy users of glycoprotein IIb/IIIa inhibitors.
Where I can see prasugrel being used prior to the cath lab at Duke is in ST-elevation MI, where the rate of PCI is very high. In primary angioplasty for ST-elevation MI, it would likely be given upstream. The bigger issue for us will be that we serve as a referral base for a lot of regional hospitals, and thus have some influence on their practices.
Dr. Bhatt: In that case, what would you advise those regional hospitals to do for non-ST-elevation MI?
Dr. Alexander: For the time being, we would advise continuing with our current practice, which is to load clopidogrel in patients in whom there is a reasonable certainty that CABG will not be performed, and to use glycoprotein IIb/IIIa inhibitors in high-risk patients. As we get more experience with prasugrel or with additional trial results, however, that practice could easily change.
Dr. Bhatt: So you would still use glycoprotein IIb/IIIa inhibitors?
Dr. Alexander: Yes, I advocate upstream clopidogrel use, but not all my colleagues do. Based on the guidelines, I’d use one or the other—either clopidogrel or a glycoprotein IIb/IIIa inhibitor. As I mentioned in my talk, if a patient is at high risk for bleeding, I am more inclined to use clopidogrel, although patients at higher risk of bleeding are often at higher risk for ischemic events as well.
WHAT’S DRIVEN THE DROPOFF IN GLYCOPROTEIN IIb/IIIa INHIBITOR USE?
Dr. Bhatt: While we’re on the topic of glycoprotein IIb/IIIa inhibitors, a question card from the audience asks why there has been a decrease in glycoprotein IIb/ IIIa inhibitor use and whether this decline is appropriate or inappropriate. Have clopidogrel pretreatment, higher loading doses of clopidogrel, and use of the direct thrombin inhibitor bivalirudin contributed to the decrease in glycoprotein IIb/IIIa inhibitor use?
Dr. Alexander: I do think that the decline has been driven by the changing environment, with greater use of other antithrombotic strategies that include clopidogrel and bivalirudin, as you suggest, as well as an increased attention to bleeding. From an evidence-based standpoint, we don’t know whether the decrease in glycoprotein IIb/IIIa use is appropriate or not because the studies of these agents were conducted before the widespread upstream use of clopidogrel and bivalirudin. Clopidogrel is attractive because it’s a pill given as one dose in the emergency department, the wards, or the catheterization laboratory, rather than a much more complicated infusion with weight-based dosing and dosage adjustments based on creatinine clearance. It is possible that we should perhaps be dosing clopidogrel the same way, but we don’t know that yet.
PRASUGREL IN PRACTICE: HOW LOW CAN THE DOSE GO, AND IS THERE A GENDER EFFECT?
Dr. Bhatt: Let’s stick with this focus on dosing but turn back to discussion of prasugrel. In your presentation of the TRITON-TIMI 38 data, Dr. Sabatine, you proposed a potential prasugrel dosage modification, down to a 5-mg loading dose, in subgroups that were identified as being at high bleeding risk—namely, elderly patients and patients with low body weight. However, no outcomes data with 5 mg of prasugrel came out of TRITON-TIMI 38.10 Is this proposed modification based on pharmacokinetic extrapolation? Could clinicians be comfortable using 5 mg of prasugrel, assuming the drug receives regulatory approval and a 5-mg tablet would be available?
Dr. Sabatine: Of course, evidence at the grade A level would consist of a trial showing that patients who received a lower dose enjoyed the same benefit as those who got standard dosing in TRITON-TIMI 38—a 60-mg loading dose followed by 10 mg/day—with an acceptable risk profile. However, such a trial would be difficult and costly to conduct, and would take roughly half a decade to pull off. It is only through large trials like TRITON-TIMI 38 that you identify subgroups that respond differently, and then to go back and do a separate trial for those subgroups takes a great deal of time. It may not be practical.
I think the Food and Drug Administration is moving toward embracing careful pharmacokinetic/pharmacodynamic substudies within trials, with these substudies having adequate numbers of subjects to provide a sense for the ideal target dose and what an acceptable dose range would be, without limiting approval to a single dose. The analogy would be warfarin dosing, with the aim being to figure out an acceptable dose range, discover which patients fall outside that range, and then model the effect of a lower dose in those patients. Thus, approving a 5-mg dose of prasugrel based on TRITON-TIMI 38 would be a reasonable approach if this dose passed muster under pharmacokinetic/pharmacodynamic modeling. If this approach were taken, there would clearly be a need for postmarketing surveillance to confirm whether the modeling on the effects of the lower dose was borne out by actual outcomes.
Dr. Bhatt: The audience has posed another interesting question raised by TRITON-TIMI 38: Can you comment on the lesser effect of prasugrel in women?
Dr. Sabatine: It is true that there was not a statistically significant effect of prasugrel among women in TRITON-TIMI 38, but statistical tests among subgroups found no significant heterogeneity for the effect between men and women, and that is the relevant measure to determine any gender effect. Keep in mind that not all subgroups represent a univariate slice of the population. For example, women generally have lower body weight than men, and since prasugrel’s net clinical benefit was reduced in patients with lower body weight, that may explain some of the differing extent of effect between men and women.
Dr. Bhatt: That’s a good point about the lack of heterogeneity between men and women. In fact, a meta-analysis of clopidogrel data conducted by one of the fellows I work with revealed that men and women appear to benefit similarly from clopidogrel.12 There was a slight signal of excess bleeding in women, but there were more elderly women in the pooled population, which may have been a confounding factor. As best as anyone can tell, antiplate-let therapy works well in both men and women.
NAVIGATING MANAGEMENT ACROSS THE SPECTRUM OF CARE
Dr. Bhatt: I would like to explore a bit further how all of these issues translate across the spectrum of care, beginning in the emergency department, which we know is a key component of the entire ACS management strategy for a health care system. What should emergency medicine doctors do, given all of the potential options—clopidogrel, different loading doses of clopidogrel, prasugrel, glycoprotein IIb/IIIa inhibitors, even bivalirudin?
Dr. Peacock: It depends on the practice setting. Some emergency physicians work at community hospitals with no backup. They must have relationships with the larger centers to which they’ll be transferring patients, because ACS patients should not be staying at community hospitals. These emergency physicians must have close relationships with the physicians who will be receiving their patients, and they know the potential head-butting with surgeons surrounding early clopidogrel use better than anybody does. If they treat with clopidogrel in the emergency room, and it turns out that the patient needs to go to the catheterization laboratory, can the receiving hospital use platelet testing to shorten the standard 5-day interval from treatment to catheterization?
Dr. Bhatt: Yes, that’s a rather useful, although not completely validated, way of using point-of-care platelet testing—to potentially reduce the time to surgery.
Dr. Peacock: Right. So if the policies for handling these types of transfer-related issues are worked out in advance, all players have a pathway to follow, which can allow quick action when necessary. If you don’t have these issues worked out in advance, you either lose many opportunities to act quickly in the emergency room or you risk taking actions that will cause problems later in the course of management.
Dr. Alexander: I totally agree. The key is to sit down with all the players involved—the surgeons, the interventional cardiologists, the intensivists, the emergency room personnel—and come up with strategies for different populations of patients. Write down the collective strategy and hang it on the wall so that everybody can be comfortable with it. The strategy can be reevaluated when prasugrel or other new antithrombotic drugs come on the market.
Dr. Peacock: The other environment is the academic center, which is even more challenging, but for different reasons. At a large academic center like the Cleveland Clinic, any of 25 different cardiologists may be taking call and receiving patients from the emergency department on a particular night. A lot of phone interaction is required to elicit the planned management strategy, including if and when the patient will be going to the cath lab. Individualizing care to a particular cardiologist then becomes a time-consuming challenge, especially in clinical situations where outcomes are time-dependent.
Dr. Alexander: Agreed. Management needs to be integrated across the entire spectrum of care. The anticoagulants that we plan to use in the cath lab will affect the antithrombotic regimen used upstream.
Dr. Kottke-Marchant: One circumstance where platelet function testing has been helpful is in determining the washout of the clopidogrel effect before surgery. At Cleveland Clinic, we have implemented platelet function testing in this circumstance instead of waiting a blanket 5 days after clopidogrel administration to go to surgery. A return to normal platelet function on platelet aggregation testing, depending on the cutoff value used, is an indicator that the patient can proceed to surgery.
Dr. Bhatt: That’s a logical approach. How should we be using antiplatelet therapy in the medically managed patient, Dr. Alexander?
Dr. Alexander: When I think of medical management, I include patients who don’t go to the cath lab, but also those who do, with regards to their management prior to and following their time in the cath lab.
In patients who don’t go to the cath lab for angiography, the ACC/AHA guidelines recommend aspirin and either clopidogrel, a glycoprotein IIb/IIIa inhibitor, or both.1 In making this choice, I consider the patient’s risk of bleeding and the dosing complexity of the regimen, especially with the use of glycoprotein IIb/IIIa inhibitors in a patient with renal insufficiency. In a patient at relatively low risk for bleeding, I often use both clopidogrel and a glycoprotein IIb/IIIa inhibitor, although this strategy does not have a lot of data to support it.
The more challenging population consists of patients who go to the cath lab but do not undergo PCI; this population is managed medically too. We often drop the ball with clopidogrel in this population. Many patients in whom PCI is not performed do not receive clopidogrel upstream, for all of the reasons we’ve discussed, and there is pretty good evidence that if clopidogrel is not instituted before hospital discharge, the patient is not likely to be receiving it at 30 days either. We have an obligation to treat these patients.
Treatment following bypass surgery is much murkier, and I don’t really know what we should be doing. The ACC/AHA guidelines suggest that clopidogrel be started in a patient with non-ST-elevation ACS after bypass surgery,1 but I believe the evidence to support that recommendation is pretty weak.
Dr. Bhatt: Well, the CURE trial did contain a sizeable group that underwent bypass surgery,7 and although this group was underpowered in some respects, it was still a very large group, so I personally favor treatment in those patients. We should mention that an ongoing trial called TRILOGY ACS is comparing clopidogrel and prasugrel specifically in patients who are being managed medically,13 so more data on this strategy will be emerging.
ARE GUIDELINES DESTINED TO BECOME EVER MORE COMPLEX?
Dr. Bhatt: Here’s a comment and question from the audience that pulls together a lot of what we’ve discussed while also looking forward: The antiplatelet therapy guidelines are already complicated. If the ongoing studies of emerging antiplatelet drugs all have results that are qualitatively similar to those of the TRITON-TIMI 38 study of prasugrel—ie, better efficacy with more potent therapy but more bleeding—how do you foresee these antiplatelet drugs being used in clinical practice?
Dr. Sabatine: The contrast between the US guidelines and the European guidelines for ACS management is stark. The US guidelines—from the ACC and AHA1—are essentially an encyclopedia that includes nearly every trial of anti-platelet therapy in ACS along with complicated algorithms; they do a wonderful job of being complete. The European guidelines14 are probably one tenth the size of their US counterpart document, and they suggest treatments for various patient types; they are very simple.
In a sense, the US guidelines lay out the data and force practitioners to evaluate the trials and consider how our patients fit into the study populations. In this way they are analogous to current guidelines for anticoagulant therapy. Several anticoagulants have been compared with heparin in clinical trials. These newer anticoagulants appear to reduce the risk of ischemic events compared with heparin; some have lower rates of bleeding, while others have higher rates of bleeding. There have been few head-to-head studies of these agents, however, so we wind up with guidelines that are not definitive but rather suggest agents to “consider” in various settings.
It’s unlikely that a head-to-head trial will be conducted comparing prasugrel with the reversible P2Y12 antagonist AZD6140, assuming that both are approved for marketing. If the drugs appear equally efficacious in placebo-controlled trials, it will take consensus to determine the appropriate choice at your hospital, factoring in your patient profile, the cost of the drugs, and other variables. It’s more complicated when one agent is slightly more efficacious but causes more bleeding or, conversely, a little less efficacious but less apt to cause bleeding. In such cases, you may need to tailor therapy to the patient, trying to gauge bleeding risk. All of the emerging data appear to point to the importance of bleeding on outcomes: patients who bleed fare poorly, in part due to the bleeding itself and in part perhaps because they have a proclivity for bleeding.
THE FUTURE: MONITORING-BASED DOSING AND NICHE ANTIPLATELETS?
Dr. Bhatt: That’s a good observation. Let’s wrap up by having the other panelists share any final thoughts you may have.
Dr. Alexander: I’d like to return to the issue of measuring antiplatelet response and using it to guide therapy. Earlier we cited the examples of antihypertensive therapy and lipid-lowering therapy to support this model of monitoring-based treatment. Guidelines for dyslipidemia treatment recommend using LDL-C levels to guide therapy, but this practice is difficult to study in a randomized trial. In fact, none of the randomized trials of statins used LDL-C levels to guide therapy. They all studied fixed doses of statins versus placebo or fixed doses of another statin. Higher doses of statins were always beneficial compared with lower doses, and this finding was extrapolated into the guidelines as a justification to treat to target LDL-C levels.
Dr. Bhatt: It’s not even necessarily clear that LDL-C level is the best target, if you consider the JUPITER trial, in which patients received statin therapy based on their baseline level of high-sensitivity C-reactive protein, not their LDL-C level.15 It goes to show how incomplete our knowledge of a class of drugs may be, even decades after the drugs are introduced.
Dr. Kottke-Marchant: To speak to Dr. Alexander’s point, dose adjustment guided by platelet monitoring is a bit more problematic for antiplatelet drugs that are irreversible inhibitors, such as clopidogrel and aspirin, than for those that are reversible inhibitors, which are being developed and may eventually make more sense to use. From a drug development standpoint, a drug that requires monitoring and dose adjustment will not gain wide acceptance because it will increase medical costs and morbidity.
Dr. Bhatt: Yes, we know from experience with warfarin that doctors and patients don’t like the ongoing need for monitoring and testing.
Dr. Peacock: The drugs that are going to be adopted by the emergency department are those with the shortest half-lives, for several reasons: (1) using a drug with a short half-life won’t commit us to a particular course of action; (2) the potential for drug interactions is lower; and (3) in the event of an erroneous diagnosis, the consequence of misapplication may be mitigated by early recognition and termination of the drug. If we later decide that we’ve gone down the wrong therapeutic road or reached a wrong diagnosis, or if a complication occurs, we can turn off the therapy quickly. That level of flexibility is needed.
Dr. Kottke-Marchant: I think we are moving into an era of niche antiplatelet drugs. One might be used in a patient going to surgery, for example, and another for long-term therapy.
Dr. Peacock: One thing that I don’t have a feel for is how to transition from one drug to another. When you change drugs for a patient, it so often seems like it goes badly. If we’re eventually going to use drugs with ultra-short half-lives in the in the emergency department for the first day or two, and then switch patients to a pill for a week, a lot more platelet function testing may be needed.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/ non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2007; 50:e1–e157.
- Bonello L, Camoin-Jau L, Arques S, et al. Adjusted clopidogrel loading doses according to vasodilator-stimulated phosphoprotein phosphorylation index decrease rate of major adverse cardiovascular events in patients with clopidogrel resistance: a multicenter randomized prospective study. J Am Coll Cardiol 2008; 51:1404–1411.
- Glauser J, Emerman CL, Bhatt DL, Peacock WF. Platelet aspirin resistance in emergency department patients with suspected acute coronary syndrome. Am J Emerg Med. In press
- Patti G, Nusca A, Mangiacapra F, Gatto L, D’Ambrosio A, Di Sciascio G. Point-of-care measurement of clopidogrel responsiveness predicts clinical outcome in patients undergoing percutaneous coronary intervention. J Am Coll Cardiol 2008; 52:1128–1133.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. J Am Coll Cardiol 2008; 52:1502–1517.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- ISIS-2 Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- EARLY-ACS: Glycoprotein IIb/IIIa inhibition in patients with non-ST-segment elevation acute coronary syndrome. Clinical Trials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00089895. Updated December 17, 2008. Accessed December 18, 2008.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wiviott SD, Braunwald E, McCabe CH, et al. Intensive oral anti-platelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary dyndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: a subanalysis of a randomised trial. Lancet 2008; 371:1353–1363.
- Berger JS, Bhatt DL, Chen Z, et al. The relationship between sex, mortality and cardiovascular events among patients with established cardiovascular disease: a meta-analysis [ACC abstract 1012-149]. J Am Coll Cardiol 2008; 51 10 suppl A:A247.
- TRILOGY ACS: A comparison of prasugrel and clopidogrel in acute coronary syndrome subjects. ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00699998. Updated December 15, 2008. Accessed January 2, 2009.
- Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
CASE STUDY: THROMBOSIS AFTER STENTING DESPITE ANTIPLATELET THERAPY
Dr. Deepak Bhatt: We have taken in a wealth of terrific information from the three preceding talks in this symposium. Let’s now share some questions from the audience and explore some of the points raised in the preceding talks in a bit more practical detail for clinicians. Our three prior speakers are joined in this panel discussion by Cleveland Clinic’s Dr. Frank Peacock, who brings an emergency medicine perspective.
Let’s begin with a case-based question supplied from the audience. The patient is a 42-year-old morbidly obese man without diabetes who had a non-ST-elevation myocardial infarction (MI) less than 1 year ago. A drug-eluting stent was placed at the time of his MI, and now restenosis has occurred. He is on aspirin and clopidogrel 75 mg/day. Do you recommend running a vasodilator-stimulated phosphoprotein (VASP) test and possibly increasing the clopidogrel dose to 150 mg/ day, or should the patient just be switched to prasugrel (assuming it is commercially available) without running the VASP test?
I’ll take a quick initial stab at this question. Studies of antiplatelet therapies to prevent instent restenosis have been a mixed bag. Some of the trials with glycoprotein IIb/IIIa inhibitors have shown an effect on restenosis, but most have not. Similarly, some of the analyses of the thienopyridines ticlopodine and clopidogrel have shown an effect on restenosis, but most have not.
For the most part, restenosis does not appear to be heavily mediated by platelets, at least not in a way that we can influence by therapy. On the other hand, stent thrombosis is highly platelet mediated, so I would alter the case to one in which stent thrombosis is the clinical problem. Assuming that the patient has been adherent to his antiplatelet regimen, which tests would you perform, and how would you act on the information from those tests?
Dr. Kandice Kottke-Marchant: The 2007 guidelines on acute coronary syndrome (ACS) management from the American College of Cardiology and American Heart Association (ACC/AHA)1 do not address platelet function testing, and almost none of the clinical trials of antiplatelet agents had an arm that included testing and dose adjustment based on platelet function studies. Platelet testing is available at some centers; at Cleveland Clinic, we use platelet aggregation testing. One can do platelet aggregation testing on a patient-by-patient basis; if inhibition appears to be suboptimal, a treatment decision should be made, but there is little guidance from the literature to steer that decision. I have seen clinicians increase the dose of clopidogrel or aspirin in response to platelet function tests, which occasionally triggers a confirmatory call from the pharmacy department.
Dr. Bhatt: When I was still at Cleveland Clinic, our chief medical resident did an analysis of platelet function testing, and it was remarkable how much testing was performed and how often it changed management, largely in the absence of any outcomes data, as Dr. Kottke-Marchant pointed out. Dr. Alexander, what are your recommendations with respect to platelet function testing today?
Dr. John Alexander: The case you describe is one in which applying evidence is not easy. There are no trials to supply any evidence to change therapy in this patient, a morbidly obese man receiving 75 mg/day of clopidogrel. There is certainly a rationale, however, to believe that a standard “one size fits all” 75-mg daily dose of clopidogrel may not be enough for him. The trade-off with a higher dosage is a higher risk of bleeding, however, so I would first be sure that he has been adherent to his current regimen of clopidogrel and aspirin.
Dr. Bhatt: Is there a role for point-of-care testing to determine whether he is adherent to the medicines?
Dr. Kottke-Marchant: Several of the point-of-care tests, such as the VerifyNow rapid platelet function analyzer, have specific cartridges for aspirin and for clopidogrel. If platelets were not being inhibited, it would suggest that the doses were too low, given the patient’s weight, but you probably would not be able to determine whether he was resistant to clopidogrel.
Dr. W. Frank Peacock: One way to verify that patients are taking their aspirin is to take a small urine sample and squirt in 2 mL of ferric chloride. If the sample turns purple, it means they are taking their aspirin. Once that is established, you can try to determine whether the drug is working on their platelets.
Dr. Alexander: Another potential explanation for stent thrombosis is faulty stent placement. In this case I would consider asking an interventional colleague to perform intravascular ultrasonography to make sure the stent was implanted properly before I changed the patient’s antithrombotic therapy.
Dr. Bhatt: That’s a great technical point. We always want to make sure that a case of stent thrombosis is not due to a mechanical problem. We should be asking: Is the stent properly sized and well opposed? Is there a distal dissection or any other issue that could predispose to stent thrombosis?
Dr. Alexander: This case illustrates a host of other challenges that underscore how much more work we need to do to define optimal antiplatelet therapy. Suppose we perform platelet function testing and find a low level of platelet inhibition in this patient with stent thrombosis, and we change his antiplatelet regimen. When should we test him again? If we retest in 3 months and find that he has a higher than expected level of platelet inhibition on the new antiplatelet regimen, do we dial down the intensity? Once again, there is no evidence to guide these decisions, and levels of platelet inhibition are driven not just by the medications but also by what is going on in the patient’s platelets—it is quite multifactorial.
POINT-OF-CARE PLATELET FUNCTION TESTING: CURRENT LIMITS, FUTURE ROLES
Dr. Bhatt: While we’re discussing platelet function testing, I found it interesting, Dr. Kottke-Marchant, that you said the use of bleeding time as a platelet test is finally going away. Testing of bleeding time has been around forever, but I agree that it doesn’t have much value in clinical practice. Do you think bleeding time will continue to have any role in drug development? Most phase 2 trials, and certainly phase 1 trials, still capture bleeding time to assess whether or not a drug is working. Should that, too, be jettisoned, or does bleeding time still have some merit in this context?
Dr. Kottke-Marchant: I would jettison it in drug development as well because of the considerable variability in bleeding time. It is not a test that can be standardized, and no quality control can be done. The results depend on skin turgor, age, and many other variables.
We need a global assay that will pick up multiple aspects of platelet function, such as flow-based adhesion, aggregation, and granule release. The bleeding time is a shear-dependent test, whereas the platelet aggregation test that is used in most drug trials is an artificial assay that measures only aggregation, but not under shear. The VerifyNow rapid platelet function analyzer does not measure platelets under shear and is not a global assay.
Dr. Marc Sabatine: I would underscore the need for a reliable point-of-care test of platelet function. When we prescribe a statin or an antihypertensive drug, we don’t just send the patient out the door and hope that everything will be okay. We measure the response, knowing that genotype, environmental factors, or medication factors can affect the response. When we prescribe an antiplatelet drug, we need a reliable point-of-care device to make certain that the patient is getting appropriate platelet inhibition.
I am reminded of a recent study of point-of-care measurement of platelet inhibition in patients receiving clopidogrel prior to nonemergent percutaneous coronary intervention (PCI).2 Rather than just treating patients with PCI and sending them out the door, the investigators kept giving patients clopidogrel and measuring their platelet inhibition until they achieved an appropriate degree of inhibition, after which PCI was performed. Event rates were significantly reduced in the patient group treated this way, which suggests a need to individualize therapy and move away from the “one size fits all” mindset.
Dr. Bhatt: Dr. Peacock, you’ve led a study of point-of-care assays in the emergency department. What might ultimately be the role of point-of-care testing in emergency medicine, and might it influence drug selection?
Dr. Peacock: My short answer is that I think there will be a role for point-of-care testing, with all the caveats that have been discussed. There may even be a day when we do genetic testing and look for DNA. Honestly, though, I’m somewhat of a skeptic because I’m not looking at the genetics. I see many patients who do crack cocaine who come to the emergency room with chest pain and have risk factors, but I send these patients home because they are not having an event. The real question is, “Is it an event?” If a patient is having an event and he or she has platelet resistance or hyperreactivity—whatever we term it—then you have to decide the next step.
As you mentioned, we just completed a study that evaluated a couple hundred patients for platelet inhibition resistance to aspirin, and one finding was that the incidence of platelet resistance to aspirin was much lower than we had anticipated. Studies from the literature suggest that the prevalence of resistance is around 30%, but in our study it was 6.5%.3
Dr. Kottke-Marchant: It depends on how and in whom you measure resistance. Different tests will give you different numbers. Even among studies using the same measurement techniques, the results depend on the patient population. If it’s a fairly stable cardiac population, you may see aspirin resistance rates of 4% or 5%. If it’s a population of patients who have had multiple MIs, the rate may be higher.
Dr. Peacock: That’s exactly my point. In the emergency department we see a mixed bag. We see many people who have had no prior events and have no acute event occurring. So in that setting you are going to get results that suggest that no intervention is required, whereas in that small percentage of patients in whom something is happening, your drug choice may be different.
Dr. Alexander: We are still talking about resistance to antiplatelet drugs as though it were a patient-level variable, but it’s my impression that it changes over time and within a patient.
Dr. Kottke-Marchant: It can change over time. There aren’t many good longitudinal studies. Most of the studies of “aspirin resistance” are really snapshot studies with measurements taken at one point in time. A term I prefer is “platelet reactivity.” To really assess treatment efficacy, we are going to have to look at the basal level of platelet reactivity.
WHAT ROLE FOR GENOTYPING IN GUIDING ANTIPLATELET THERAPY?
Dr. Bhatt: Dr. Peacock alluded to a potential role for genetic testing. Dr. Sabatine, you have done a lot of interesting work with genotyping in the TRITON-TIMI 38 study of prasugrel and clopidogrel. What is the future role of genotyping in determining which antiplatelet therapy is best for which patient?
Dr. Sabatine: As I mentioned, cytochrome P450 enzymes play a critical role in the metabolism of clopidogrel. These enzymes are fairly polymorphic—mutations in their encoding genes are responsible for subtle changes in effect, unlike the traditional mutations that we think about for sickle cell disease, for example. A wealth of data has been published showing that genetic variants are associated with decreased functional activity of cytochrome P450 enzymes, demonstrating the pharmacologic importance of these variants.
Individuals who carry variant alleles appear to respond differently to clopidogrel. A variety of small studies show that those who carry specific variants—particularly in the CYP2C19 enzyme, but in other enzymes as well—appear to have a diminished response to clopidogrel. There are also data showing that individuals with a diminished response to clopidogrel have worse outcomes.4 Our group is studying the impact of genetic variants that decrease the functional activity of cytochrome P450 enzymes on clinical outcomes. (Editor’s note: This study has since been published by Mega et al.5)
The practical implication may lie in point-of-care genotyping, which appears possible and will be clinically useful if a strong link can be demonstrated between genotype and outcomes. If point-of-care genotyping becomes practical, it will raise the question of whether both genotyping and platelet aggregation testing are needed. I think they might indeed be complementary in risk prediction, as is the case with genetic variants that affect low-density lipoprotein cholesterol (LDL-C) levels. In the lipid arena, we have seen that genetic effects and lipid levels provide independent incremental information about risk. That’s because of the high degree of variation in LDL-C levels: an LDL-C measurement is a snapshot in time, yet a variety of factors can influence LDL-C levels. In contrast, genotype is an invariant factor. Similarly, in the platelet arena, platelet aggregation studies and genotyping may be synergistic in predicting an individual’s predisposition to events and response to medications.
Dr. Bhatt: While we’re discussing pathways of metabolism, the literature, though scant, suggests a potential interaction between proton pump inhibitors and clopidogrel. I was co-chair of a recent American College of Cardiology/ American Heart Association/American College of Gastro-enterology consensus document that endorsed liberal use of proton pump inhibitors in patients who are at gastrointestinal risk, including those on antiplatelet therapy.6 The gastroenterologists believed strongly that proton pump inhibitors were safe and in fact underused in these patients. What do you think about the clopidogrel–proton pump inhibitor interaction? Should we be concerned?
Dr. Sabatine: Proton pump inhibitors are not only substrates for, but also inhibitors of, CYP2C19, a key enzyme that helps transform clopidogrel into an active metabolite. For this reason, there has been interest in whether concomitant use of proton pump inhibitors would blunt the efficacy of clopidogrel. The same concern was raised about giving clopidogrel with certain statin drugs that are also metabolized by the cytochrome P450 system, and several studies have shown an effect of these statins on clopidogrel’s platelet inhibition. However, there is no evidence that coadministration of these statins has affected clinical outcomes with clopidogrel in clinical trials. So it may be that while competition for the cytochrome P450 system is one factor, it’s not enough of a factor to tip the scale and result in a clinical event. The same may be true of coadministration of proton pump inhibitors; meanwhile, we await definitive data that concomitant use with clopidogrel leads to higher rates of ischemic events.
DIAGNOSTIC UNCERTAINTY IN THE EMERGENCY SETTING
Dr. Bhatt: We heard about quite a few new antiplatelet drugs in Dr. Sabatine’s presentation, some of which will likely be taken up in clinical practice. Dr. Peacock, from an emergency department perspective, how will you integrate all these new agents with the numerous therapies already available? What should emergency departments do to come to grips with and ultimately take advantage of these different forms of therapy as well as emerging platelet function tests?
Dr. Peacock: The piece that’s unique or especially pertinent to the emergency department is diagnostic uncertainty. Diagnosis and management are easy when a patient has an ST-elevation MI because we all know what that looks like and we know what to do in response. To some extent non-ST-elevation MI is fairly simple too. ACS is a lot more difficult because we don’t have a good definition for unstable angina, and that’s where diagnosis and management become problematic. And with high-sensitivity troponins coming out now, the question of non-ST-elevation MI is going to get more and more confusing because we will have a lot more patients who meet criteria without having an acute coronary artery event.
So it is going to be important that studies be designed correctly. A lot of the studies reviewed today were efficacy studies, showing that a particular drug works well in a carefully defined population, but they were not efficiency studies: they did not take into account the real-world diagnostic uncertainty—and inevitable misdiagnoses—that emergency departments encounter before starting therapy.
Take the CURE trial, for example. It was a great study, showing that clopidogrel reduced the hazard ratio for major coronary events by 20% in patients with unstable angina,7 and the message was that everybody should be using clopidogrel. A close look at the study, however, reveals that about half the patients did not receive clopidogrel in the emergency department. When patients did receive it early, it was driven by the cardiologist, who was absolutely certain of the diagnosis. But if the study was not designed to test early use, then it is a big leap to extrapolate its findings to this circumstance.
Many of the patients in the CURE trial were enrolled the day after presentation, when their diagnosis was certain—ie, they had a rise in troponin after their symptoms. But when a patient first arrives in the emergency department, we are not certain of the diagnosis. And if we use a drug such as clopidogrel, with a duration of action as long as 5 days, we have committed the entire medical system to a certain course of management for that period of time. If we get the diagnosis wrong, this commitment could restrict management options for up to 5 days.
The question for emergency physicians becomes, “How long is long enough to know whether I can pull the trigger on a therapy and be correct?” With all the new drugs coming along, the way to answer this is to do efficiency studies in a real-world environment in addition to efficacy studies.
Dr. Alexander: Yes, one of the biggest limitations of antiplatelet drug studies to date is that they usually haven’t really addressed the timing of drug initiation. We often assume that if a drug is shown to be beneficial, then it should be started as soon as possible. As we just heard, that may have been an unfounded extrapolation from the CURE trial. The same sort of thing happened with the ISIS trial of aspirin in patients with ST-elevation MI.8 In response to the ISIS results, clinicians rushed to give patients aspirin right away even though many of the patients in the trial may have received their aspirin the day after presentation. For these reasons, the EARLY-ACS study,9 which is addressing a very simple question—whether early upstream use of glycoprotein IIb/IIIa inhibitors is beneficial—has been a challenging trial to complete.
WHAT ROLE FOR THIENOPYRIDINE PRETREATMENT?
Dr. Bhatt: Dr. Sabatine, you presented data from the large TRITON-TIMI 38 trial comparing prasugrel with clopidogrel. I’m interested in how you would use prasugrel in practice, assuming it receives marketing approval, especially in light of its bleeding risk, particularly in patients in whom coronary artery bypass graft surgery (CABG) is planned. Many hospitals pretreat patients with clopidogrel in the emergency department. How would you manage a patient who shows up in the emergency room with ACS? Would you give clopidogrel, would you wait and give prasugrel, or would you do something else? If you gave clopidogrel, what loading dose would you use—300 mg, 600 mg, or, as some have suggested, 900 or 1,200 mg?
Dr. Sabatine: I am a strong proponent of pretreatment. Data from multiple studies show a benefit to this strategy, and even the original CURE trial showed a roughly 30% reduction in ischemic events within the first 24 hours of clopidogrel initiation.7
I think the dosing strategy depends on how the patient is going to be managed. If management is going to be conservative, then I would start the patient on 300 mg of clopidogrel when he or she came in. If the patient is going to the cardiac catheterization laboratory in a few hours, I would pretreat with 600 mg of clopidogrel. For prasugrel, the need for pretreatment is less clear, given the drug’s faster onset of action and greater degree of platelet inhibition. In the TRITON-TIMI 38 study,10 prasugrel was given, by and large, after diagnostic angiography, and thus one could use that approach in practice.
In terms of clopidogrel versus prasugrel, I would embrace prasugrel for the large majority of my patients, being mindful of the risk of bleeding. I would not hesitate to give the medication to diabetics or to younger, more robust patients. The 50% reduction in stent thrombosis with prasugrel versus clopidogrel in TRITON-TIMI 38 is huge,11 given that the risk of death with stent thrombosis is probably 25% or greater. So I would want to have prasugrel on board to reduce the risk of stent thrombosis, especially if a drug-eluting stent were being implanted.
Dr. Bhatt: Dr. Alexander, let’s get your take on a similar scenario. Assuming that prasugrel gains marketing approval, how would you manage patients with non-ST-elevation MI who present to the emergency department? Would you pretreat with clopidogrel? Would you wait until angiography and then, depending on the anatomy, treat with prasugrel? Or would you potentially pretreat with prasugrel, which has not been studied and would not be a labeled indication? How would you reconcile the data?
Dr. Alexander: At Duke, I expect that prasugrel will not be used prior to the catheterization laboratory in patients with non-ST-elevation ACS due to concerns about whether the patients will undergo PCI or be managed medically or with CABG.
Dr. Bhatt: That makes sense, since there was a fair amount of bleeding with prasugrel in those patients in TRITON-TIMI 38.
Dr. Alexander: Correct. Moreover, at Duke we don’t use as much upstream clopidogrel as we would, based on the evidence, if I were managing all the patients. There is still a lot of pushback about upstream clopidogrel from our surgeons because patients are going to surgery quickly these days, sometimes just a day after catheterization, and that’s when a loading dose of clopidogrel can be problematic. We are also still fairly heavy users of glycoprotein IIb/IIIa inhibitors.
Where I can see prasugrel being used prior to the cath lab at Duke is in ST-elevation MI, where the rate of PCI is very high. In primary angioplasty for ST-elevation MI, it would likely be given upstream. The bigger issue for us will be that we serve as a referral base for a lot of regional hospitals, and thus have some influence on their practices.
Dr. Bhatt: In that case, what would you advise those regional hospitals to do for non-ST-elevation MI?
Dr. Alexander: For the time being, we would advise continuing with our current practice, which is to load clopidogrel in patients in whom there is a reasonable certainty that CABG will not be performed, and to use glycoprotein IIb/IIIa inhibitors in high-risk patients. As we get more experience with prasugrel or with additional trial results, however, that practice could easily change.
Dr. Bhatt: So you would still use glycoprotein IIb/IIIa inhibitors?
Dr. Alexander: Yes, I advocate upstream clopidogrel use, but not all my colleagues do. Based on the guidelines, I’d use one or the other—either clopidogrel or a glycoprotein IIb/IIIa inhibitor. As I mentioned in my talk, if a patient is at high risk for bleeding, I am more inclined to use clopidogrel, although patients at higher risk of bleeding are often at higher risk for ischemic events as well.
WHAT’S DRIVEN THE DROPOFF IN GLYCOPROTEIN IIb/IIIa INHIBITOR USE?
Dr. Bhatt: While we’re on the topic of glycoprotein IIb/IIIa inhibitors, a question card from the audience asks why there has been a decrease in glycoprotein IIb/ IIIa inhibitor use and whether this decline is appropriate or inappropriate. Have clopidogrel pretreatment, higher loading doses of clopidogrel, and use of the direct thrombin inhibitor bivalirudin contributed to the decrease in glycoprotein IIb/IIIa inhibitor use?
Dr. Alexander: I do think that the decline has been driven by the changing environment, with greater use of other antithrombotic strategies that include clopidogrel and bivalirudin, as you suggest, as well as an increased attention to bleeding. From an evidence-based standpoint, we don’t know whether the decrease in glycoprotein IIb/IIIa use is appropriate or not because the studies of these agents were conducted before the widespread upstream use of clopidogrel and bivalirudin. Clopidogrel is attractive because it’s a pill given as one dose in the emergency department, the wards, or the catheterization laboratory, rather than a much more complicated infusion with weight-based dosing and dosage adjustments based on creatinine clearance. It is possible that we should perhaps be dosing clopidogrel the same way, but we don’t know that yet.
PRASUGREL IN PRACTICE: HOW LOW CAN THE DOSE GO, AND IS THERE A GENDER EFFECT?
Dr. Bhatt: Let’s stick with this focus on dosing but turn back to discussion of prasugrel. In your presentation of the TRITON-TIMI 38 data, Dr. Sabatine, you proposed a potential prasugrel dosage modification, down to a 5-mg loading dose, in subgroups that were identified as being at high bleeding risk—namely, elderly patients and patients with low body weight. However, no outcomes data with 5 mg of prasugrel came out of TRITON-TIMI 38.10 Is this proposed modification based on pharmacokinetic extrapolation? Could clinicians be comfortable using 5 mg of prasugrel, assuming the drug receives regulatory approval and a 5-mg tablet would be available?
Dr. Sabatine: Of course, evidence at the grade A level would consist of a trial showing that patients who received a lower dose enjoyed the same benefit as those who got standard dosing in TRITON-TIMI 38—a 60-mg loading dose followed by 10 mg/day—with an acceptable risk profile. However, such a trial would be difficult and costly to conduct, and would take roughly half a decade to pull off. It is only through large trials like TRITON-TIMI 38 that you identify subgroups that respond differently, and then to go back and do a separate trial for those subgroups takes a great deal of time. It may not be practical.
I think the Food and Drug Administration is moving toward embracing careful pharmacokinetic/pharmacodynamic substudies within trials, with these substudies having adequate numbers of subjects to provide a sense for the ideal target dose and what an acceptable dose range would be, without limiting approval to a single dose. The analogy would be warfarin dosing, with the aim being to figure out an acceptable dose range, discover which patients fall outside that range, and then model the effect of a lower dose in those patients. Thus, approving a 5-mg dose of prasugrel based on TRITON-TIMI 38 would be a reasonable approach if this dose passed muster under pharmacokinetic/pharmacodynamic modeling. If this approach were taken, there would clearly be a need for postmarketing surveillance to confirm whether the modeling on the effects of the lower dose was borne out by actual outcomes.
Dr. Bhatt: The audience has posed another interesting question raised by TRITON-TIMI 38: Can you comment on the lesser effect of prasugrel in women?
Dr. Sabatine: It is true that there was not a statistically significant effect of prasugrel among women in TRITON-TIMI 38, but statistical tests among subgroups found no significant heterogeneity for the effect between men and women, and that is the relevant measure to determine any gender effect. Keep in mind that not all subgroups represent a univariate slice of the population. For example, women generally have lower body weight than men, and since prasugrel’s net clinical benefit was reduced in patients with lower body weight, that may explain some of the differing extent of effect between men and women.
Dr. Bhatt: That’s a good point about the lack of heterogeneity between men and women. In fact, a meta-analysis of clopidogrel data conducted by one of the fellows I work with revealed that men and women appear to benefit similarly from clopidogrel.12 There was a slight signal of excess bleeding in women, but there were more elderly women in the pooled population, which may have been a confounding factor. As best as anyone can tell, antiplate-let therapy works well in both men and women.
NAVIGATING MANAGEMENT ACROSS THE SPECTRUM OF CARE
Dr. Bhatt: I would like to explore a bit further how all of these issues translate across the spectrum of care, beginning in the emergency department, which we know is a key component of the entire ACS management strategy for a health care system. What should emergency medicine doctors do, given all of the potential options—clopidogrel, different loading doses of clopidogrel, prasugrel, glycoprotein IIb/IIIa inhibitors, even bivalirudin?
Dr. Peacock: It depends on the practice setting. Some emergency physicians work at community hospitals with no backup. They must have relationships with the larger centers to which they’ll be transferring patients, because ACS patients should not be staying at community hospitals. These emergency physicians must have close relationships with the physicians who will be receiving their patients, and they know the potential head-butting with surgeons surrounding early clopidogrel use better than anybody does. If they treat with clopidogrel in the emergency room, and it turns out that the patient needs to go to the catheterization laboratory, can the receiving hospital use platelet testing to shorten the standard 5-day interval from treatment to catheterization?
Dr. Bhatt: Yes, that’s a rather useful, although not completely validated, way of using point-of-care platelet testing—to potentially reduce the time to surgery.
Dr. Peacock: Right. So if the policies for handling these types of transfer-related issues are worked out in advance, all players have a pathway to follow, which can allow quick action when necessary. If you don’t have these issues worked out in advance, you either lose many opportunities to act quickly in the emergency room or you risk taking actions that will cause problems later in the course of management.
Dr. Alexander: I totally agree. The key is to sit down with all the players involved—the surgeons, the interventional cardiologists, the intensivists, the emergency room personnel—and come up with strategies for different populations of patients. Write down the collective strategy and hang it on the wall so that everybody can be comfortable with it. The strategy can be reevaluated when prasugrel or other new antithrombotic drugs come on the market.
Dr. Peacock: The other environment is the academic center, which is even more challenging, but for different reasons. At a large academic center like the Cleveland Clinic, any of 25 different cardiologists may be taking call and receiving patients from the emergency department on a particular night. A lot of phone interaction is required to elicit the planned management strategy, including if and when the patient will be going to the cath lab. Individualizing care to a particular cardiologist then becomes a time-consuming challenge, especially in clinical situations where outcomes are time-dependent.
Dr. Alexander: Agreed. Management needs to be integrated across the entire spectrum of care. The anticoagulants that we plan to use in the cath lab will affect the antithrombotic regimen used upstream.
Dr. Kottke-Marchant: One circumstance where platelet function testing has been helpful is in determining the washout of the clopidogrel effect before surgery. At Cleveland Clinic, we have implemented platelet function testing in this circumstance instead of waiting a blanket 5 days after clopidogrel administration to go to surgery. A return to normal platelet function on platelet aggregation testing, depending on the cutoff value used, is an indicator that the patient can proceed to surgery.
Dr. Bhatt: That’s a logical approach. How should we be using antiplatelet therapy in the medically managed patient, Dr. Alexander?
Dr. Alexander: When I think of medical management, I include patients who don’t go to the cath lab, but also those who do, with regards to their management prior to and following their time in the cath lab.
In patients who don’t go to the cath lab for angiography, the ACC/AHA guidelines recommend aspirin and either clopidogrel, a glycoprotein IIb/IIIa inhibitor, or both.1 In making this choice, I consider the patient’s risk of bleeding and the dosing complexity of the regimen, especially with the use of glycoprotein IIb/IIIa inhibitors in a patient with renal insufficiency. In a patient at relatively low risk for bleeding, I often use both clopidogrel and a glycoprotein IIb/IIIa inhibitor, although this strategy does not have a lot of data to support it.
The more challenging population consists of patients who go to the cath lab but do not undergo PCI; this population is managed medically too. We often drop the ball with clopidogrel in this population. Many patients in whom PCI is not performed do not receive clopidogrel upstream, for all of the reasons we’ve discussed, and there is pretty good evidence that if clopidogrel is not instituted before hospital discharge, the patient is not likely to be receiving it at 30 days either. We have an obligation to treat these patients.
Treatment following bypass surgery is much murkier, and I don’t really know what we should be doing. The ACC/AHA guidelines suggest that clopidogrel be started in a patient with non-ST-elevation ACS after bypass surgery,1 but I believe the evidence to support that recommendation is pretty weak.
Dr. Bhatt: Well, the CURE trial did contain a sizeable group that underwent bypass surgery,7 and although this group was underpowered in some respects, it was still a very large group, so I personally favor treatment in those patients. We should mention that an ongoing trial called TRILOGY ACS is comparing clopidogrel and prasugrel specifically in patients who are being managed medically,13 so more data on this strategy will be emerging.
ARE GUIDELINES DESTINED TO BECOME EVER MORE COMPLEX?
Dr. Bhatt: Here’s a comment and question from the audience that pulls together a lot of what we’ve discussed while also looking forward: The antiplatelet therapy guidelines are already complicated. If the ongoing studies of emerging antiplatelet drugs all have results that are qualitatively similar to those of the TRITON-TIMI 38 study of prasugrel—ie, better efficacy with more potent therapy but more bleeding—how do you foresee these antiplatelet drugs being used in clinical practice?
Dr. Sabatine: The contrast between the US guidelines and the European guidelines for ACS management is stark. The US guidelines—from the ACC and AHA1—are essentially an encyclopedia that includes nearly every trial of anti-platelet therapy in ACS along with complicated algorithms; they do a wonderful job of being complete. The European guidelines14 are probably one tenth the size of their US counterpart document, and they suggest treatments for various patient types; they are very simple.
In a sense, the US guidelines lay out the data and force practitioners to evaluate the trials and consider how our patients fit into the study populations. In this way they are analogous to current guidelines for anticoagulant therapy. Several anticoagulants have been compared with heparin in clinical trials. These newer anticoagulants appear to reduce the risk of ischemic events compared with heparin; some have lower rates of bleeding, while others have higher rates of bleeding. There have been few head-to-head studies of these agents, however, so we wind up with guidelines that are not definitive but rather suggest agents to “consider” in various settings.
It’s unlikely that a head-to-head trial will be conducted comparing prasugrel with the reversible P2Y12 antagonist AZD6140, assuming that both are approved for marketing. If the drugs appear equally efficacious in placebo-controlled trials, it will take consensus to determine the appropriate choice at your hospital, factoring in your patient profile, the cost of the drugs, and other variables. It’s more complicated when one agent is slightly more efficacious but causes more bleeding or, conversely, a little less efficacious but less apt to cause bleeding. In such cases, you may need to tailor therapy to the patient, trying to gauge bleeding risk. All of the emerging data appear to point to the importance of bleeding on outcomes: patients who bleed fare poorly, in part due to the bleeding itself and in part perhaps because they have a proclivity for bleeding.
THE FUTURE: MONITORING-BASED DOSING AND NICHE ANTIPLATELETS?
Dr. Bhatt: That’s a good observation. Let’s wrap up by having the other panelists share any final thoughts you may have.
Dr. Alexander: I’d like to return to the issue of measuring antiplatelet response and using it to guide therapy. Earlier we cited the examples of antihypertensive therapy and lipid-lowering therapy to support this model of monitoring-based treatment. Guidelines for dyslipidemia treatment recommend using LDL-C levels to guide therapy, but this practice is difficult to study in a randomized trial. In fact, none of the randomized trials of statins used LDL-C levels to guide therapy. They all studied fixed doses of statins versus placebo or fixed doses of another statin. Higher doses of statins were always beneficial compared with lower doses, and this finding was extrapolated into the guidelines as a justification to treat to target LDL-C levels.
Dr. Bhatt: It’s not even necessarily clear that LDL-C level is the best target, if you consider the JUPITER trial, in which patients received statin therapy based on their baseline level of high-sensitivity C-reactive protein, not their LDL-C level.15 It goes to show how incomplete our knowledge of a class of drugs may be, even decades after the drugs are introduced.
Dr. Kottke-Marchant: To speak to Dr. Alexander’s point, dose adjustment guided by platelet monitoring is a bit more problematic for antiplatelet drugs that are irreversible inhibitors, such as clopidogrel and aspirin, than for those that are reversible inhibitors, which are being developed and may eventually make more sense to use. From a drug development standpoint, a drug that requires monitoring and dose adjustment will not gain wide acceptance because it will increase medical costs and morbidity.
Dr. Bhatt: Yes, we know from experience with warfarin that doctors and patients don’t like the ongoing need for monitoring and testing.
Dr. Peacock: The drugs that are going to be adopted by the emergency department are those with the shortest half-lives, for several reasons: (1) using a drug with a short half-life won’t commit us to a particular course of action; (2) the potential for drug interactions is lower; and (3) in the event of an erroneous diagnosis, the consequence of misapplication may be mitigated by early recognition and termination of the drug. If we later decide that we’ve gone down the wrong therapeutic road or reached a wrong diagnosis, or if a complication occurs, we can turn off the therapy quickly. That level of flexibility is needed.
Dr. Kottke-Marchant: I think we are moving into an era of niche antiplatelet drugs. One might be used in a patient going to surgery, for example, and another for long-term therapy.
Dr. Peacock: One thing that I don’t have a feel for is how to transition from one drug to another. When you change drugs for a patient, it so often seems like it goes badly. If we’re eventually going to use drugs with ultra-short half-lives in the in the emergency department for the first day or two, and then switch patients to a pill for a week, a lot more platelet function testing may be needed.
CASE STUDY: THROMBOSIS AFTER STENTING DESPITE ANTIPLATELET THERAPY
Dr. Deepak Bhatt: We have taken in a wealth of terrific information from the three preceding talks in this symposium. Let’s now share some questions from the audience and explore some of the points raised in the preceding talks in a bit more practical detail for clinicians. Our three prior speakers are joined in this panel discussion by Cleveland Clinic’s Dr. Frank Peacock, who brings an emergency medicine perspective.
Let’s begin with a case-based question supplied from the audience. The patient is a 42-year-old morbidly obese man without diabetes who had a non-ST-elevation myocardial infarction (MI) less than 1 year ago. A drug-eluting stent was placed at the time of his MI, and now restenosis has occurred. He is on aspirin and clopidogrel 75 mg/day. Do you recommend running a vasodilator-stimulated phosphoprotein (VASP) test and possibly increasing the clopidogrel dose to 150 mg/ day, or should the patient just be switched to prasugrel (assuming it is commercially available) without running the VASP test?
I’ll take a quick initial stab at this question. Studies of antiplatelet therapies to prevent instent restenosis have been a mixed bag. Some of the trials with glycoprotein IIb/IIIa inhibitors have shown an effect on restenosis, but most have not. Similarly, some of the analyses of the thienopyridines ticlopodine and clopidogrel have shown an effect on restenosis, but most have not.
For the most part, restenosis does not appear to be heavily mediated by platelets, at least not in a way that we can influence by therapy. On the other hand, stent thrombosis is highly platelet mediated, so I would alter the case to one in which stent thrombosis is the clinical problem. Assuming that the patient has been adherent to his antiplatelet regimen, which tests would you perform, and how would you act on the information from those tests?
Dr. Kandice Kottke-Marchant: The 2007 guidelines on acute coronary syndrome (ACS) management from the American College of Cardiology and American Heart Association (ACC/AHA)1 do not address platelet function testing, and almost none of the clinical trials of antiplatelet agents had an arm that included testing and dose adjustment based on platelet function studies. Platelet testing is available at some centers; at Cleveland Clinic, we use platelet aggregation testing. One can do platelet aggregation testing on a patient-by-patient basis; if inhibition appears to be suboptimal, a treatment decision should be made, but there is little guidance from the literature to steer that decision. I have seen clinicians increase the dose of clopidogrel or aspirin in response to platelet function tests, which occasionally triggers a confirmatory call from the pharmacy department.
Dr. Bhatt: When I was still at Cleveland Clinic, our chief medical resident did an analysis of platelet function testing, and it was remarkable how much testing was performed and how often it changed management, largely in the absence of any outcomes data, as Dr. Kottke-Marchant pointed out. Dr. Alexander, what are your recommendations with respect to platelet function testing today?
Dr. John Alexander: The case you describe is one in which applying evidence is not easy. There are no trials to supply any evidence to change therapy in this patient, a morbidly obese man receiving 75 mg/day of clopidogrel. There is certainly a rationale, however, to believe that a standard “one size fits all” 75-mg daily dose of clopidogrel may not be enough for him. The trade-off with a higher dosage is a higher risk of bleeding, however, so I would first be sure that he has been adherent to his current regimen of clopidogrel and aspirin.
Dr. Bhatt: Is there a role for point-of-care testing to determine whether he is adherent to the medicines?
Dr. Kottke-Marchant: Several of the point-of-care tests, such as the VerifyNow rapid platelet function analyzer, have specific cartridges for aspirin and for clopidogrel. If platelets were not being inhibited, it would suggest that the doses were too low, given the patient’s weight, but you probably would not be able to determine whether he was resistant to clopidogrel.
Dr. W. Frank Peacock: One way to verify that patients are taking their aspirin is to take a small urine sample and squirt in 2 mL of ferric chloride. If the sample turns purple, it means they are taking their aspirin. Once that is established, you can try to determine whether the drug is working on their platelets.
Dr. Alexander: Another potential explanation for stent thrombosis is faulty stent placement. In this case I would consider asking an interventional colleague to perform intravascular ultrasonography to make sure the stent was implanted properly before I changed the patient’s antithrombotic therapy.
Dr. Bhatt: That’s a great technical point. We always want to make sure that a case of stent thrombosis is not due to a mechanical problem. We should be asking: Is the stent properly sized and well opposed? Is there a distal dissection or any other issue that could predispose to stent thrombosis?
Dr. Alexander: This case illustrates a host of other challenges that underscore how much more work we need to do to define optimal antiplatelet therapy. Suppose we perform platelet function testing and find a low level of platelet inhibition in this patient with stent thrombosis, and we change his antiplatelet regimen. When should we test him again? If we retest in 3 months and find that he has a higher than expected level of platelet inhibition on the new antiplatelet regimen, do we dial down the intensity? Once again, there is no evidence to guide these decisions, and levels of platelet inhibition are driven not just by the medications but also by what is going on in the patient’s platelets—it is quite multifactorial.
POINT-OF-CARE PLATELET FUNCTION TESTING: CURRENT LIMITS, FUTURE ROLES
Dr. Bhatt: While we’re discussing platelet function testing, I found it interesting, Dr. Kottke-Marchant, that you said the use of bleeding time as a platelet test is finally going away. Testing of bleeding time has been around forever, but I agree that it doesn’t have much value in clinical practice. Do you think bleeding time will continue to have any role in drug development? Most phase 2 trials, and certainly phase 1 trials, still capture bleeding time to assess whether or not a drug is working. Should that, too, be jettisoned, or does bleeding time still have some merit in this context?
Dr. Kottke-Marchant: I would jettison it in drug development as well because of the considerable variability in bleeding time. It is not a test that can be standardized, and no quality control can be done. The results depend on skin turgor, age, and many other variables.
We need a global assay that will pick up multiple aspects of platelet function, such as flow-based adhesion, aggregation, and granule release. The bleeding time is a shear-dependent test, whereas the platelet aggregation test that is used in most drug trials is an artificial assay that measures only aggregation, but not under shear. The VerifyNow rapid platelet function analyzer does not measure platelets under shear and is not a global assay.
Dr. Marc Sabatine: I would underscore the need for a reliable point-of-care test of platelet function. When we prescribe a statin or an antihypertensive drug, we don’t just send the patient out the door and hope that everything will be okay. We measure the response, knowing that genotype, environmental factors, or medication factors can affect the response. When we prescribe an antiplatelet drug, we need a reliable point-of-care device to make certain that the patient is getting appropriate platelet inhibition.
I am reminded of a recent study of point-of-care measurement of platelet inhibition in patients receiving clopidogrel prior to nonemergent percutaneous coronary intervention (PCI).2 Rather than just treating patients with PCI and sending them out the door, the investigators kept giving patients clopidogrel and measuring their platelet inhibition until they achieved an appropriate degree of inhibition, after which PCI was performed. Event rates were significantly reduced in the patient group treated this way, which suggests a need to individualize therapy and move away from the “one size fits all” mindset.
Dr. Bhatt: Dr. Peacock, you’ve led a study of point-of-care assays in the emergency department. What might ultimately be the role of point-of-care testing in emergency medicine, and might it influence drug selection?
Dr. Peacock: My short answer is that I think there will be a role for point-of-care testing, with all the caveats that have been discussed. There may even be a day when we do genetic testing and look for DNA. Honestly, though, I’m somewhat of a skeptic because I’m not looking at the genetics. I see many patients who do crack cocaine who come to the emergency room with chest pain and have risk factors, but I send these patients home because they are not having an event. The real question is, “Is it an event?” If a patient is having an event and he or she has platelet resistance or hyperreactivity—whatever we term it—then you have to decide the next step.
As you mentioned, we just completed a study that evaluated a couple hundred patients for platelet inhibition resistance to aspirin, and one finding was that the incidence of platelet resistance to aspirin was much lower than we had anticipated. Studies from the literature suggest that the prevalence of resistance is around 30%, but in our study it was 6.5%.3
Dr. Kottke-Marchant: It depends on how and in whom you measure resistance. Different tests will give you different numbers. Even among studies using the same measurement techniques, the results depend on the patient population. If it’s a fairly stable cardiac population, you may see aspirin resistance rates of 4% or 5%. If it’s a population of patients who have had multiple MIs, the rate may be higher.
Dr. Peacock: That’s exactly my point. In the emergency department we see a mixed bag. We see many people who have had no prior events and have no acute event occurring. So in that setting you are going to get results that suggest that no intervention is required, whereas in that small percentage of patients in whom something is happening, your drug choice may be different.
Dr. Alexander: We are still talking about resistance to antiplatelet drugs as though it were a patient-level variable, but it’s my impression that it changes over time and within a patient.
Dr. Kottke-Marchant: It can change over time. There aren’t many good longitudinal studies. Most of the studies of “aspirin resistance” are really snapshot studies with measurements taken at one point in time. A term I prefer is “platelet reactivity.” To really assess treatment efficacy, we are going to have to look at the basal level of platelet reactivity.
WHAT ROLE FOR GENOTYPING IN GUIDING ANTIPLATELET THERAPY?
Dr. Bhatt: Dr. Peacock alluded to a potential role for genetic testing. Dr. Sabatine, you have done a lot of interesting work with genotyping in the TRITON-TIMI 38 study of prasugrel and clopidogrel. What is the future role of genotyping in determining which antiplatelet therapy is best for which patient?
Dr. Sabatine: As I mentioned, cytochrome P450 enzymes play a critical role in the metabolism of clopidogrel. These enzymes are fairly polymorphic—mutations in their encoding genes are responsible for subtle changes in effect, unlike the traditional mutations that we think about for sickle cell disease, for example. A wealth of data has been published showing that genetic variants are associated with decreased functional activity of cytochrome P450 enzymes, demonstrating the pharmacologic importance of these variants.
Individuals who carry variant alleles appear to respond differently to clopidogrel. A variety of small studies show that those who carry specific variants—particularly in the CYP2C19 enzyme, but in other enzymes as well—appear to have a diminished response to clopidogrel. There are also data showing that individuals with a diminished response to clopidogrel have worse outcomes.4 Our group is studying the impact of genetic variants that decrease the functional activity of cytochrome P450 enzymes on clinical outcomes. (Editor’s note: This study has since been published by Mega et al.5)
The practical implication may lie in point-of-care genotyping, which appears possible and will be clinically useful if a strong link can be demonstrated between genotype and outcomes. If point-of-care genotyping becomes practical, it will raise the question of whether both genotyping and platelet aggregation testing are needed. I think they might indeed be complementary in risk prediction, as is the case with genetic variants that affect low-density lipoprotein cholesterol (LDL-C) levels. In the lipid arena, we have seen that genetic effects and lipid levels provide independent incremental information about risk. That’s because of the high degree of variation in LDL-C levels: an LDL-C measurement is a snapshot in time, yet a variety of factors can influence LDL-C levels. In contrast, genotype is an invariant factor. Similarly, in the platelet arena, platelet aggregation studies and genotyping may be synergistic in predicting an individual’s predisposition to events and response to medications.
Dr. Bhatt: While we’re discussing pathways of metabolism, the literature, though scant, suggests a potential interaction between proton pump inhibitors and clopidogrel. I was co-chair of a recent American College of Cardiology/ American Heart Association/American College of Gastro-enterology consensus document that endorsed liberal use of proton pump inhibitors in patients who are at gastrointestinal risk, including those on antiplatelet therapy.6 The gastroenterologists believed strongly that proton pump inhibitors were safe and in fact underused in these patients. What do you think about the clopidogrel–proton pump inhibitor interaction? Should we be concerned?
Dr. Sabatine: Proton pump inhibitors are not only substrates for, but also inhibitors of, CYP2C19, a key enzyme that helps transform clopidogrel into an active metabolite. For this reason, there has been interest in whether concomitant use of proton pump inhibitors would blunt the efficacy of clopidogrel. The same concern was raised about giving clopidogrel with certain statin drugs that are also metabolized by the cytochrome P450 system, and several studies have shown an effect of these statins on clopidogrel’s platelet inhibition. However, there is no evidence that coadministration of these statins has affected clinical outcomes with clopidogrel in clinical trials. So it may be that while competition for the cytochrome P450 system is one factor, it’s not enough of a factor to tip the scale and result in a clinical event. The same may be true of coadministration of proton pump inhibitors; meanwhile, we await definitive data that concomitant use with clopidogrel leads to higher rates of ischemic events.
DIAGNOSTIC UNCERTAINTY IN THE EMERGENCY SETTING
Dr. Bhatt: We heard about quite a few new antiplatelet drugs in Dr. Sabatine’s presentation, some of which will likely be taken up in clinical practice. Dr. Peacock, from an emergency department perspective, how will you integrate all these new agents with the numerous therapies already available? What should emergency departments do to come to grips with and ultimately take advantage of these different forms of therapy as well as emerging platelet function tests?
Dr. Peacock: The piece that’s unique or especially pertinent to the emergency department is diagnostic uncertainty. Diagnosis and management are easy when a patient has an ST-elevation MI because we all know what that looks like and we know what to do in response. To some extent non-ST-elevation MI is fairly simple too. ACS is a lot more difficult because we don’t have a good definition for unstable angina, and that’s where diagnosis and management become problematic. And with high-sensitivity troponins coming out now, the question of non-ST-elevation MI is going to get more and more confusing because we will have a lot more patients who meet criteria without having an acute coronary artery event.
So it is going to be important that studies be designed correctly. A lot of the studies reviewed today were efficacy studies, showing that a particular drug works well in a carefully defined population, but they were not efficiency studies: they did not take into account the real-world diagnostic uncertainty—and inevitable misdiagnoses—that emergency departments encounter before starting therapy.
Take the CURE trial, for example. It was a great study, showing that clopidogrel reduced the hazard ratio for major coronary events by 20% in patients with unstable angina,7 and the message was that everybody should be using clopidogrel. A close look at the study, however, reveals that about half the patients did not receive clopidogrel in the emergency department. When patients did receive it early, it was driven by the cardiologist, who was absolutely certain of the diagnosis. But if the study was not designed to test early use, then it is a big leap to extrapolate its findings to this circumstance.
Many of the patients in the CURE trial were enrolled the day after presentation, when their diagnosis was certain—ie, they had a rise in troponin after their symptoms. But when a patient first arrives in the emergency department, we are not certain of the diagnosis. And if we use a drug such as clopidogrel, with a duration of action as long as 5 days, we have committed the entire medical system to a certain course of management for that period of time. If we get the diagnosis wrong, this commitment could restrict management options for up to 5 days.
The question for emergency physicians becomes, “How long is long enough to know whether I can pull the trigger on a therapy and be correct?” With all the new drugs coming along, the way to answer this is to do efficiency studies in a real-world environment in addition to efficacy studies.
Dr. Alexander: Yes, one of the biggest limitations of antiplatelet drug studies to date is that they usually haven’t really addressed the timing of drug initiation. We often assume that if a drug is shown to be beneficial, then it should be started as soon as possible. As we just heard, that may have been an unfounded extrapolation from the CURE trial. The same sort of thing happened with the ISIS trial of aspirin in patients with ST-elevation MI.8 In response to the ISIS results, clinicians rushed to give patients aspirin right away even though many of the patients in the trial may have received their aspirin the day after presentation. For these reasons, the EARLY-ACS study,9 which is addressing a very simple question—whether early upstream use of glycoprotein IIb/IIIa inhibitors is beneficial—has been a challenging trial to complete.
WHAT ROLE FOR THIENOPYRIDINE PRETREATMENT?
Dr. Bhatt: Dr. Sabatine, you presented data from the large TRITON-TIMI 38 trial comparing prasugrel with clopidogrel. I’m interested in how you would use prasugrel in practice, assuming it receives marketing approval, especially in light of its bleeding risk, particularly in patients in whom coronary artery bypass graft surgery (CABG) is planned. Many hospitals pretreat patients with clopidogrel in the emergency department. How would you manage a patient who shows up in the emergency room with ACS? Would you give clopidogrel, would you wait and give prasugrel, or would you do something else? If you gave clopidogrel, what loading dose would you use—300 mg, 600 mg, or, as some have suggested, 900 or 1,200 mg?
Dr. Sabatine: I am a strong proponent of pretreatment. Data from multiple studies show a benefit to this strategy, and even the original CURE trial showed a roughly 30% reduction in ischemic events within the first 24 hours of clopidogrel initiation.7
I think the dosing strategy depends on how the patient is going to be managed. If management is going to be conservative, then I would start the patient on 300 mg of clopidogrel when he or she came in. If the patient is going to the cardiac catheterization laboratory in a few hours, I would pretreat with 600 mg of clopidogrel. For prasugrel, the need for pretreatment is less clear, given the drug’s faster onset of action and greater degree of platelet inhibition. In the TRITON-TIMI 38 study,10 prasugrel was given, by and large, after diagnostic angiography, and thus one could use that approach in practice.
In terms of clopidogrel versus prasugrel, I would embrace prasugrel for the large majority of my patients, being mindful of the risk of bleeding. I would not hesitate to give the medication to diabetics or to younger, more robust patients. The 50% reduction in stent thrombosis with prasugrel versus clopidogrel in TRITON-TIMI 38 is huge,11 given that the risk of death with stent thrombosis is probably 25% or greater. So I would want to have prasugrel on board to reduce the risk of stent thrombosis, especially if a drug-eluting stent were being implanted.
Dr. Bhatt: Dr. Alexander, let’s get your take on a similar scenario. Assuming that prasugrel gains marketing approval, how would you manage patients with non-ST-elevation MI who present to the emergency department? Would you pretreat with clopidogrel? Would you wait until angiography and then, depending on the anatomy, treat with prasugrel? Or would you potentially pretreat with prasugrel, which has not been studied and would not be a labeled indication? How would you reconcile the data?
Dr. Alexander: At Duke, I expect that prasugrel will not be used prior to the catheterization laboratory in patients with non-ST-elevation ACS due to concerns about whether the patients will undergo PCI or be managed medically or with CABG.
Dr. Bhatt: That makes sense, since there was a fair amount of bleeding with prasugrel in those patients in TRITON-TIMI 38.
Dr. Alexander: Correct. Moreover, at Duke we don’t use as much upstream clopidogrel as we would, based on the evidence, if I were managing all the patients. There is still a lot of pushback about upstream clopidogrel from our surgeons because patients are going to surgery quickly these days, sometimes just a day after catheterization, and that’s when a loading dose of clopidogrel can be problematic. We are also still fairly heavy users of glycoprotein IIb/IIIa inhibitors.
Where I can see prasugrel being used prior to the cath lab at Duke is in ST-elevation MI, where the rate of PCI is very high. In primary angioplasty for ST-elevation MI, it would likely be given upstream. The bigger issue for us will be that we serve as a referral base for a lot of regional hospitals, and thus have some influence on their practices.
Dr. Bhatt: In that case, what would you advise those regional hospitals to do for non-ST-elevation MI?
Dr. Alexander: For the time being, we would advise continuing with our current practice, which is to load clopidogrel in patients in whom there is a reasonable certainty that CABG will not be performed, and to use glycoprotein IIb/IIIa inhibitors in high-risk patients. As we get more experience with prasugrel or with additional trial results, however, that practice could easily change.
Dr. Bhatt: So you would still use glycoprotein IIb/IIIa inhibitors?
Dr. Alexander: Yes, I advocate upstream clopidogrel use, but not all my colleagues do. Based on the guidelines, I’d use one or the other—either clopidogrel or a glycoprotein IIb/IIIa inhibitor. As I mentioned in my talk, if a patient is at high risk for bleeding, I am more inclined to use clopidogrel, although patients at higher risk of bleeding are often at higher risk for ischemic events as well.
WHAT’S DRIVEN THE DROPOFF IN GLYCOPROTEIN IIb/IIIa INHIBITOR USE?
Dr. Bhatt: While we’re on the topic of glycoprotein IIb/IIIa inhibitors, a question card from the audience asks why there has been a decrease in glycoprotein IIb/ IIIa inhibitor use and whether this decline is appropriate or inappropriate. Have clopidogrel pretreatment, higher loading doses of clopidogrel, and use of the direct thrombin inhibitor bivalirudin contributed to the decrease in glycoprotein IIb/IIIa inhibitor use?
Dr. Alexander: I do think that the decline has been driven by the changing environment, with greater use of other antithrombotic strategies that include clopidogrel and bivalirudin, as you suggest, as well as an increased attention to bleeding. From an evidence-based standpoint, we don’t know whether the decrease in glycoprotein IIb/IIIa use is appropriate or not because the studies of these agents were conducted before the widespread upstream use of clopidogrel and bivalirudin. Clopidogrel is attractive because it’s a pill given as one dose in the emergency department, the wards, or the catheterization laboratory, rather than a much more complicated infusion with weight-based dosing and dosage adjustments based on creatinine clearance. It is possible that we should perhaps be dosing clopidogrel the same way, but we don’t know that yet.
PRASUGREL IN PRACTICE: HOW LOW CAN THE DOSE GO, AND IS THERE A GENDER EFFECT?
Dr. Bhatt: Let’s stick with this focus on dosing but turn back to discussion of prasugrel. In your presentation of the TRITON-TIMI 38 data, Dr. Sabatine, you proposed a potential prasugrel dosage modification, down to a 5-mg loading dose, in subgroups that were identified as being at high bleeding risk—namely, elderly patients and patients with low body weight. However, no outcomes data with 5 mg of prasugrel came out of TRITON-TIMI 38.10 Is this proposed modification based on pharmacokinetic extrapolation? Could clinicians be comfortable using 5 mg of prasugrel, assuming the drug receives regulatory approval and a 5-mg tablet would be available?
Dr. Sabatine: Of course, evidence at the grade A level would consist of a trial showing that patients who received a lower dose enjoyed the same benefit as those who got standard dosing in TRITON-TIMI 38—a 60-mg loading dose followed by 10 mg/day—with an acceptable risk profile. However, such a trial would be difficult and costly to conduct, and would take roughly half a decade to pull off. It is only through large trials like TRITON-TIMI 38 that you identify subgroups that respond differently, and then to go back and do a separate trial for those subgroups takes a great deal of time. It may not be practical.
I think the Food and Drug Administration is moving toward embracing careful pharmacokinetic/pharmacodynamic substudies within trials, with these substudies having adequate numbers of subjects to provide a sense for the ideal target dose and what an acceptable dose range would be, without limiting approval to a single dose. The analogy would be warfarin dosing, with the aim being to figure out an acceptable dose range, discover which patients fall outside that range, and then model the effect of a lower dose in those patients. Thus, approving a 5-mg dose of prasugrel based on TRITON-TIMI 38 would be a reasonable approach if this dose passed muster under pharmacokinetic/pharmacodynamic modeling. If this approach were taken, there would clearly be a need for postmarketing surveillance to confirm whether the modeling on the effects of the lower dose was borne out by actual outcomes.
Dr. Bhatt: The audience has posed another interesting question raised by TRITON-TIMI 38: Can you comment on the lesser effect of prasugrel in women?
Dr. Sabatine: It is true that there was not a statistically significant effect of prasugrel among women in TRITON-TIMI 38, but statistical tests among subgroups found no significant heterogeneity for the effect between men and women, and that is the relevant measure to determine any gender effect. Keep in mind that not all subgroups represent a univariate slice of the population. For example, women generally have lower body weight than men, and since prasugrel’s net clinical benefit was reduced in patients with lower body weight, that may explain some of the differing extent of effect between men and women.
Dr. Bhatt: That’s a good point about the lack of heterogeneity between men and women. In fact, a meta-analysis of clopidogrel data conducted by one of the fellows I work with revealed that men and women appear to benefit similarly from clopidogrel.12 There was a slight signal of excess bleeding in women, but there were more elderly women in the pooled population, which may have been a confounding factor. As best as anyone can tell, antiplate-let therapy works well in both men and women.
NAVIGATING MANAGEMENT ACROSS THE SPECTRUM OF CARE
Dr. Bhatt: I would like to explore a bit further how all of these issues translate across the spectrum of care, beginning in the emergency department, which we know is a key component of the entire ACS management strategy for a health care system. What should emergency medicine doctors do, given all of the potential options—clopidogrel, different loading doses of clopidogrel, prasugrel, glycoprotein IIb/IIIa inhibitors, even bivalirudin?
Dr. Peacock: It depends on the practice setting. Some emergency physicians work at community hospitals with no backup. They must have relationships with the larger centers to which they’ll be transferring patients, because ACS patients should not be staying at community hospitals. These emergency physicians must have close relationships with the physicians who will be receiving their patients, and they know the potential head-butting with surgeons surrounding early clopidogrel use better than anybody does. If they treat with clopidogrel in the emergency room, and it turns out that the patient needs to go to the catheterization laboratory, can the receiving hospital use platelet testing to shorten the standard 5-day interval from treatment to catheterization?
Dr. Bhatt: Yes, that’s a rather useful, although not completely validated, way of using point-of-care platelet testing—to potentially reduce the time to surgery.
Dr. Peacock: Right. So if the policies for handling these types of transfer-related issues are worked out in advance, all players have a pathway to follow, which can allow quick action when necessary. If you don’t have these issues worked out in advance, you either lose many opportunities to act quickly in the emergency room or you risk taking actions that will cause problems later in the course of management.
Dr. Alexander: I totally agree. The key is to sit down with all the players involved—the surgeons, the interventional cardiologists, the intensivists, the emergency room personnel—and come up with strategies for different populations of patients. Write down the collective strategy and hang it on the wall so that everybody can be comfortable with it. The strategy can be reevaluated when prasugrel or other new antithrombotic drugs come on the market.
Dr. Peacock: The other environment is the academic center, which is even more challenging, but for different reasons. At a large academic center like the Cleveland Clinic, any of 25 different cardiologists may be taking call and receiving patients from the emergency department on a particular night. A lot of phone interaction is required to elicit the planned management strategy, including if and when the patient will be going to the cath lab. Individualizing care to a particular cardiologist then becomes a time-consuming challenge, especially in clinical situations where outcomes are time-dependent.
Dr. Alexander: Agreed. Management needs to be integrated across the entire spectrum of care. The anticoagulants that we plan to use in the cath lab will affect the antithrombotic regimen used upstream.
Dr. Kottke-Marchant: One circumstance where platelet function testing has been helpful is in determining the washout of the clopidogrel effect before surgery. At Cleveland Clinic, we have implemented platelet function testing in this circumstance instead of waiting a blanket 5 days after clopidogrel administration to go to surgery. A return to normal platelet function on platelet aggregation testing, depending on the cutoff value used, is an indicator that the patient can proceed to surgery.
Dr. Bhatt: That’s a logical approach. How should we be using antiplatelet therapy in the medically managed patient, Dr. Alexander?
Dr. Alexander: When I think of medical management, I include patients who don’t go to the cath lab, but also those who do, with regards to their management prior to and following their time in the cath lab.
In patients who don’t go to the cath lab for angiography, the ACC/AHA guidelines recommend aspirin and either clopidogrel, a glycoprotein IIb/IIIa inhibitor, or both.1 In making this choice, I consider the patient’s risk of bleeding and the dosing complexity of the regimen, especially with the use of glycoprotein IIb/IIIa inhibitors in a patient with renal insufficiency. In a patient at relatively low risk for bleeding, I often use both clopidogrel and a glycoprotein IIb/IIIa inhibitor, although this strategy does not have a lot of data to support it.
The more challenging population consists of patients who go to the cath lab but do not undergo PCI; this population is managed medically too. We often drop the ball with clopidogrel in this population. Many patients in whom PCI is not performed do not receive clopidogrel upstream, for all of the reasons we’ve discussed, and there is pretty good evidence that if clopidogrel is not instituted before hospital discharge, the patient is not likely to be receiving it at 30 days either. We have an obligation to treat these patients.
Treatment following bypass surgery is much murkier, and I don’t really know what we should be doing. The ACC/AHA guidelines suggest that clopidogrel be started in a patient with non-ST-elevation ACS after bypass surgery,1 but I believe the evidence to support that recommendation is pretty weak.
Dr. Bhatt: Well, the CURE trial did contain a sizeable group that underwent bypass surgery,7 and although this group was underpowered in some respects, it was still a very large group, so I personally favor treatment in those patients. We should mention that an ongoing trial called TRILOGY ACS is comparing clopidogrel and prasugrel specifically in patients who are being managed medically,13 so more data on this strategy will be emerging.
ARE GUIDELINES DESTINED TO BECOME EVER MORE COMPLEX?
Dr. Bhatt: Here’s a comment and question from the audience that pulls together a lot of what we’ve discussed while also looking forward: The antiplatelet therapy guidelines are already complicated. If the ongoing studies of emerging antiplatelet drugs all have results that are qualitatively similar to those of the TRITON-TIMI 38 study of prasugrel—ie, better efficacy with more potent therapy but more bleeding—how do you foresee these antiplatelet drugs being used in clinical practice?
Dr. Sabatine: The contrast between the US guidelines and the European guidelines for ACS management is stark. The US guidelines—from the ACC and AHA1—are essentially an encyclopedia that includes nearly every trial of anti-platelet therapy in ACS along with complicated algorithms; they do a wonderful job of being complete. The European guidelines14 are probably one tenth the size of their US counterpart document, and they suggest treatments for various patient types; they are very simple.
In a sense, the US guidelines lay out the data and force practitioners to evaluate the trials and consider how our patients fit into the study populations. In this way they are analogous to current guidelines for anticoagulant therapy. Several anticoagulants have been compared with heparin in clinical trials. These newer anticoagulants appear to reduce the risk of ischemic events compared with heparin; some have lower rates of bleeding, while others have higher rates of bleeding. There have been few head-to-head studies of these agents, however, so we wind up with guidelines that are not definitive but rather suggest agents to “consider” in various settings.
It’s unlikely that a head-to-head trial will be conducted comparing prasugrel with the reversible P2Y12 antagonist AZD6140, assuming that both are approved for marketing. If the drugs appear equally efficacious in placebo-controlled trials, it will take consensus to determine the appropriate choice at your hospital, factoring in your patient profile, the cost of the drugs, and other variables. It’s more complicated when one agent is slightly more efficacious but causes more bleeding or, conversely, a little less efficacious but less apt to cause bleeding. In such cases, you may need to tailor therapy to the patient, trying to gauge bleeding risk. All of the emerging data appear to point to the importance of bleeding on outcomes: patients who bleed fare poorly, in part due to the bleeding itself and in part perhaps because they have a proclivity for bleeding.
THE FUTURE: MONITORING-BASED DOSING AND NICHE ANTIPLATELETS?
Dr. Bhatt: That’s a good observation. Let’s wrap up by having the other panelists share any final thoughts you may have.
Dr. Alexander: I’d like to return to the issue of measuring antiplatelet response and using it to guide therapy. Earlier we cited the examples of antihypertensive therapy and lipid-lowering therapy to support this model of monitoring-based treatment. Guidelines for dyslipidemia treatment recommend using LDL-C levels to guide therapy, but this practice is difficult to study in a randomized trial. In fact, none of the randomized trials of statins used LDL-C levels to guide therapy. They all studied fixed doses of statins versus placebo or fixed doses of another statin. Higher doses of statins were always beneficial compared with lower doses, and this finding was extrapolated into the guidelines as a justification to treat to target LDL-C levels.
Dr. Bhatt: It’s not even necessarily clear that LDL-C level is the best target, if you consider the JUPITER trial, in which patients received statin therapy based on their baseline level of high-sensitivity C-reactive protein, not their LDL-C level.15 It goes to show how incomplete our knowledge of a class of drugs may be, even decades after the drugs are introduced.
Dr. Kottke-Marchant: To speak to Dr. Alexander’s point, dose adjustment guided by platelet monitoring is a bit more problematic for antiplatelet drugs that are irreversible inhibitors, such as clopidogrel and aspirin, than for those that are reversible inhibitors, which are being developed and may eventually make more sense to use. From a drug development standpoint, a drug that requires monitoring and dose adjustment will not gain wide acceptance because it will increase medical costs and morbidity.
Dr. Bhatt: Yes, we know from experience with warfarin that doctors and patients don’t like the ongoing need for monitoring and testing.
Dr. Peacock: The drugs that are going to be adopted by the emergency department are those with the shortest half-lives, for several reasons: (1) using a drug with a short half-life won’t commit us to a particular course of action; (2) the potential for drug interactions is lower; and (3) in the event of an erroneous diagnosis, the consequence of misapplication may be mitigated by early recognition and termination of the drug. If we later decide that we’ve gone down the wrong therapeutic road or reached a wrong diagnosis, or if a complication occurs, we can turn off the therapy quickly. That level of flexibility is needed.
Dr. Kottke-Marchant: I think we are moving into an era of niche antiplatelet drugs. One might be used in a patient going to surgery, for example, and another for long-term therapy.
Dr. Peacock: One thing that I don’t have a feel for is how to transition from one drug to another. When you change drugs for a patient, it so often seems like it goes badly. If we’re eventually going to use drugs with ultra-short half-lives in the in the emergency department for the first day or two, and then switch patients to a pill for a week, a lot more platelet function testing may be needed.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/ non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2007; 50:e1–e157.
- Bonello L, Camoin-Jau L, Arques S, et al. Adjusted clopidogrel loading doses according to vasodilator-stimulated phosphoprotein phosphorylation index decrease rate of major adverse cardiovascular events in patients with clopidogrel resistance: a multicenter randomized prospective study. J Am Coll Cardiol 2008; 51:1404–1411.
- Glauser J, Emerman CL, Bhatt DL, Peacock WF. Platelet aspirin resistance in emergency department patients with suspected acute coronary syndrome. Am J Emerg Med. In press
- Patti G, Nusca A, Mangiacapra F, Gatto L, D’Ambrosio A, Di Sciascio G. Point-of-care measurement of clopidogrel responsiveness predicts clinical outcome in patients undergoing percutaneous coronary intervention. J Am Coll Cardiol 2008; 52:1128–1133.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. J Am Coll Cardiol 2008; 52:1502–1517.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- ISIS-2 Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- EARLY-ACS: Glycoprotein IIb/IIIa inhibition in patients with non-ST-segment elevation acute coronary syndrome. Clinical Trials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00089895. Updated December 17, 2008. Accessed December 18, 2008.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wiviott SD, Braunwald E, McCabe CH, et al. Intensive oral anti-platelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary dyndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: a subanalysis of a randomised trial. Lancet 2008; 371:1353–1363.
- Berger JS, Bhatt DL, Chen Z, et al. The relationship between sex, mortality and cardiovascular events among patients with established cardiovascular disease: a meta-analysis [ACC abstract 1012-149]. J Am Coll Cardiol 2008; 51 10 suppl A:A247.
- TRILOGY ACS: A comparison of prasugrel and clopidogrel in acute coronary syndrome subjects. ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00699998. Updated December 15, 2008. Accessed January 2, 2009.
- Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/ non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2007; 50:e1–e157.
- Bonello L, Camoin-Jau L, Arques S, et al. Adjusted clopidogrel loading doses according to vasodilator-stimulated phosphoprotein phosphorylation index decrease rate of major adverse cardiovascular events in patients with clopidogrel resistance: a multicenter randomized prospective study. J Am Coll Cardiol 2008; 51:1404–1411.
- Glauser J, Emerman CL, Bhatt DL, Peacock WF. Platelet aspirin resistance in emergency department patients with suspected acute coronary syndrome. Am J Emerg Med. In press
- Patti G, Nusca A, Mangiacapra F, Gatto L, D’Ambrosio A, Di Sciascio G. Point-of-care measurement of clopidogrel responsiveness predicts clinical outcome in patients undergoing percutaneous coronary intervention. J Am Coll Cardiol 2008; 52:1128–1133.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362.
- Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. J Am Coll Cardiol 2008; 52:1502–1517.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- ISIS-2 Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- EARLY-ACS: Glycoprotein IIb/IIIa inhibition in patients with non-ST-segment elevation acute coronary syndrome. Clinical Trials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00089895. Updated December 17, 2008. Accessed December 18, 2008.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wiviott SD, Braunwald E, McCabe CH, et al. Intensive oral anti-platelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary dyndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: a subanalysis of a randomised trial. Lancet 2008; 371:1353–1363.
- Berger JS, Bhatt DL, Chen Z, et al. The relationship between sex, mortality and cardiovascular events among patients with established cardiovascular disease: a meta-analysis [ACC abstract 1012-149]. J Am Coll Cardiol 2008; 51 10 suppl A:A247.
- TRILOGY ACS: A comparison of prasugrel and clopidogrel in acute coronary syndrome subjects. ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT00699998. Updated December 15, 2008. Accessed January 2, 2009.
- Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.