Article
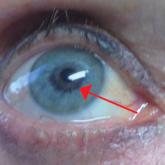
Bimatoprost-Induced Iris Hyperpigmentation: Beauty in the Darkened Eye of the Beholder
Iris hyperpigmentation can occur when bimatoprost eye drops are applied to the eyes for treatment of ocular hypertension and glaucoma, but reports...
Article
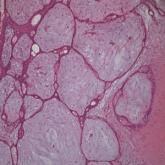
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculoma and trichodiscoma are adnexal tumors that arise from or around hair follicles and are two of the many characteristic features of...
Article
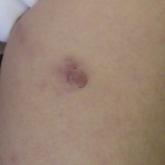
A Rare Case of Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
Primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) is a rare, intermediately aggressive form of primary cutaneous B-cell lymphoma...
