User login
Can patients opt to turn off implantable cardioverter-defibrillators near the end of life?
Yes. Although implantable cardioverter-defibrillators (ICDs) prevent sudden cardiac death in patients with advanced heart failure, their benefit in terminally ill patients is small.1 Furthermore, the shocks they deliver at the end of life can cause distress. Therefore, it is reasonable to consider ICD deactivation if the patient or family wishes.
A DIFFICULT DECISION
End-of-life decisions place significant emotional burdens on patients, their families, and their healthcare providers and can have social and legal consequences.
Turning off an ICD is an especially difficult decision, considering that these devices protect against sudden cardiac death and fatal arrhythmias. Also, patients and their representatives may find it more difficult to withdraw from active care than to forgo further interventions (more on this below), and they may misunderstand discussions about ICD deactivation, perceiving them as the beginning of abandonment.
ICD DEACTIVATION IS OFTEN DONE HAPHAZARDLY OR NOT AT ALL
Many healthcare providers are not trained in or comfortable with discussing end-of-life issues, and many hospitals and hospice programs lack policies and protocols for managing implanted devices at the end of life. Consequently, ICD management at the end of life varies among providers and tends to be suboptimal.2
In a report of a survey in 414 hospice facilities, 97% of facilities reported that they admitted patients with ICDs, but only 10% had a policy on device deactivation.3
In a survey of 47 European medical centers, only 4% said they addressed ICD deactivation with their patients.4
A study of 125 patients with ICDs who had died found that 52% had do-not-resuscitate orders. Nevertheless, in 100 patients the ICD had remained active in the last 24 hours of their life, and 31 of these patients had received shocks during their last 24 hours.5
In a survey of next of kin of patients with ICDs who had died of any cause,6 in only 27 of 100 cases had the clinician discussed ICD deactivation, and about three-fourths of these discussions had occurred during the last few days of life. Twenty-seven patients had received ICD discharges in the last month of life, and 8% had received a discharge during the final minutes.
TRAINING AND PROTOCOLS ARE NEEDED
Healthcare professionals need education about device deactivation at the end of life so that they are comfortable communicating with patients and families about this critical issue. To this end, several cardiac and palliative care societies have jointly released an expert statement on managing ICDs and other implantable devices in end-of-life situations.7
Many providers harbor a misunderstanding of the difference between withholding a device and withdrawing (or turning off) a device that is already implanted.2 Some mistakenly believe they would be committing a crime by deactivating an implanted life-sustaining device. Legally and ethically, there is no difference between withholding a device and withdrawing a device. Legally, carrying out a request to withdraw life-sustaining treatment is neither physician-assisted suicide nor euthanasia.
DISCUSSION SHOULD BEGIN EARLY AND SHOULD BE ONGOING
The discussion of ICD deactivation should begin before the device is implanted and should continue as the patient’s health status changes. In a survey, 40% of patients said they felt that ICD deactivation should be discussed before the device is implanted, and only 5% felt that this discussion should be undertaken in the last days of life.8
At the least, it is important to identify patients with ICDs on admission to hospice and to have policies in place that ensure adequate patient education to make an informed decision about ICD deactivation at the end of life.
The topic should be discussed when goals of care change and when do-not-resuscitate status is addressed, and also when advanced directives are being acknowledged. If the patient or his or her legal representative wishes to keep the ICD turned on, that wish should be respected. The essence of a discussion is not to impose the providers’ choice on the patient, but to help the patient make the right decision for himself or herself. Of note, patients entering hospice do not have to have do-not-resuscitate status.
We believe that device management in end-of-life circumstances should be part of the discussion of the goals of care. Accordingly, healthcare providers need to be familiar with device management and to have a higher comfort level in addressing such sensitive topics with patients facing the end of life, as well as with their families.
It is also advisable to apply protocols within hospice services to address ICD management options for the patient and the legal representative. An early decision regarding end-of-life deactivation will help patients avoid distressing ICD discharges and the related emotional distress in their last moments.
- Barsheshet A, Moss AJ, Huang DT, McNitt S, Zareba W, Goldenberg I. Applicability of a risk score for prediction of the long-term (8-year) benefit of the implantable cardioverter-defibrillator. J Am Coll Cardiol 2012; 59:2075–2079.
- Kapa S, Mueller PS, Hayes DL, Asirvatham SJ. Perspectives on withdrawing pacemaker and implantable cardioverter-defibrillator therapies at end of life: results of a survey of medical and legal professionals and patients. Mayo Clin Proc 2010; 85:981–990.
- Goldstein N, Carlson M, Livote E, Kutner JS. Brief communication: management of implantable cardioverter-defibrillators in hospice: a nationwide survey. Ann Intern Med 2010; 152:296–299.
- Marinskis G, van Erven L; EHRA Scientific Initiatives Committtee. Deactivation of implanted cardioverter-defibrillators at the end of life: results of the EHRA survey. Europace 2010; 12:1176–1177.
- Kinch Westerdahl A, Sjoblom J, Mattiasson AC, Rosenqvist M, Frykman V. Implantable cardioverter-defibrillator therapy before death: high risk for painful shocks at end of life. Circulation 2014; 129:422–429.
- Goldstein NE, Lampert R, Bradley E, Lynn J, Krumholz HM. Management of implantable cardioverter defibrillators in end-of-life care. Ann Intern Med 2004; 141:835–838.
- Lampert R, Hayes DL, Annas GJ, et al; American College of Cardiology; American Geriatrics Society; American Academy of Hospice and Palliative Medicine; American Heart Association; European Heart Rhythm Association; Hospice and Palliative Nurses Association. HRS expert consensus statement on the management of cardiovascular implantable electronic devices (CIEDs) in patients nearing end of life or requesting withdrawal of therapy. Heart Rhythm 2010; 7:1008–1026.
- Raphael CE, Koa-Wing M, Stain N, Wright I, Francis DP, Kanagaratnam P. Implantable cardioverter-defibrillator recipient attitudes towards device activation: how much do patients want to know? Pacing Clin Electrophysiol 2011; 34:1628–1633.
Yes. Although implantable cardioverter-defibrillators (ICDs) prevent sudden cardiac death in patients with advanced heart failure, their benefit in terminally ill patients is small.1 Furthermore, the shocks they deliver at the end of life can cause distress. Therefore, it is reasonable to consider ICD deactivation if the patient or family wishes.
A DIFFICULT DECISION
End-of-life decisions place significant emotional burdens on patients, their families, and their healthcare providers and can have social and legal consequences.
Turning off an ICD is an especially difficult decision, considering that these devices protect against sudden cardiac death and fatal arrhythmias. Also, patients and their representatives may find it more difficult to withdraw from active care than to forgo further interventions (more on this below), and they may misunderstand discussions about ICD deactivation, perceiving them as the beginning of abandonment.
ICD DEACTIVATION IS OFTEN DONE HAPHAZARDLY OR NOT AT ALL
Many healthcare providers are not trained in or comfortable with discussing end-of-life issues, and many hospitals and hospice programs lack policies and protocols for managing implanted devices at the end of life. Consequently, ICD management at the end of life varies among providers and tends to be suboptimal.2
In a report of a survey in 414 hospice facilities, 97% of facilities reported that they admitted patients with ICDs, but only 10% had a policy on device deactivation.3
In a survey of 47 European medical centers, only 4% said they addressed ICD deactivation with their patients.4
A study of 125 patients with ICDs who had died found that 52% had do-not-resuscitate orders. Nevertheless, in 100 patients the ICD had remained active in the last 24 hours of their life, and 31 of these patients had received shocks during their last 24 hours.5
In a survey of next of kin of patients with ICDs who had died of any cause,6 in only 27 of 100 cases had the clinician discussed ICD deactivation, and about three-fourths of these discussions had occurred during the last few days of life. Twenty-seven patients had received ICD discharges in the last month of life, and 8% had received a discharge during the final minutes.
TRAINING AND PROTOCOLS ARE NEEDED
Healthcare professionals need education about device deactivation at the end of life so that they are comfortable communicating with patients and families about this critical issue. To this end, several cardiac and palliative care societies have jointly released an expert statement on managing ICDs and other implantable devices in end-of-life situations.7
Many providers harbor a misunderstanding of the difference between withholding a device and withdrawing (or turning off) a device that is already implanted.2 Some mistakenly believe they would be committing a crime by deactivating an implanted life-sustaining device. Legally and ethically, there is no difference between withholding a device and withdrawing a device. Legally, carrying out a request to withdraw life-sustaining treatment is neither physician-assisted suicide nor euthanasia.
DISCUSSION SHOULD BEGIN EARLY AND SHOULD BE ONGOING
The discussion of ICD deactivation should begin before the device is implanted and should continue as the patient’s health status changes. In a survey, 40% of patients said they felt that ICD deactivation should be discussed before the device is implanted, and only 5% felt that this discussion should be undertaken in the last days of life.8
At the least, it is important to identify patients with ICDs on admission to hospice and to have policies in place that ensure adequate patient education to make an informed decision about ICD deactivation at the end of life.
The topic should be discussed when goals of care change and when do-not-resuscitate status is addressed, and also when advanced directives are being acknowledged. If the patient or his or her legal representative wishes to keep the ICD turned on, that wish should be respected. The essence of a discussion is not to impose the providers’ choice on the patient, but to help the patient make the right decision for himself or herself. Of note, patients entering hospice do not have to have do-not-resuscitate status.
We believe that device management in end-of-life circumstances should be part of the discussion of the goals of care. Accordingly, healthcare providers need to be familiar with device management and to have a higher comfort level in addressing such sensitive topics with patients facing the end of life, as well as with their families.
It is also advisable to apply protocols within hospice services to address ICD management options for the patient and the legal representative. An early decision regarding end-of-life deactivation will help patients avoid distressing ICD discharges and the related emotional distress in their last moments.
Yes. Although implantable cardioverter-defibrillators (ICDs) prevent sudden cardiac death in patients with advanced heart failure, their benefit in terminally ill patients is small.1 Furthermore, the shocks they deliver at the end of life can cause distress. Therefore, it is reasonable to consider ICD deactivation if the patient or family wishes.
A DIFFICULT DECISION
End-of-life decisions place significant emotional burdens on patients, their families, and their healthcare providers and can have social and legal consequences.
Turning off an ICD is an especially difficult decision, considering that these devices protect against sudden cardiac death and fatal arrhythmias. Also, patients and their representatives may find it more difficult to withdraw from active care than to forgo further interventions (more on this below), and they may misunderstand discussions about ICD deactivation, perceiving them as the beginning of abandonment.
ICD DEACTIVATION IS OFTEN DONE HAPHAZARDLY OR NOT AT ALL
Many healthcare providers are not trained in or comfortable with discussing end-of-life issues, and many hospitals and hospice programs lack policies and protocols for managing implanted devices at the end of life. Consequently, ICD management at the end of life varies among providers and tends to be suboptimal.2
In a report of a survey in 414 hospice facilities, 97% of facilities reported that they admitted patients with ICDs, but only 10% had a policy on device deactivation.3
In a survey of 47 European medical centers, only 4% said they addressed ICD deactivation with their patients.4
A study of 125 patients with ICDs who had died found that 52% had do-not-resuscitate orders. Nevertheless, in 100 patients the ICD had remained active in the last 24 hours of their life, and 31 of these patients had received shocks during their last 24 hours.5
In a survey of next of kin of patients with ICDs who had died of any cause,6 in only 27 of 100 cases had the clinician discussed ICD deactivation, and about three-fourths of these discussions had occurred during the last few days of life. Twenty-seven patients had received ICD discharges in the last month of life, and 8% had received a discharge during the final minutes.
TRAINING AND PROTOCOLS ARE NEEDED
Healthcare professionals need education about device deactivation at the end of life so that they are comfortable communicating with patients and families about this critical issue. To this end, several cardiac and palliative care societies have jointly released an expert statement on managing ICDs and other implantable devices in end-of-life situations.7
Many providers harbor a misunderstanding of the difference between withholding a device and withdrawing (or turning off) a device that is already implanted.2 Some mistakenly believe they would be committing a crime by deactivating an implanted life-sustaining device. Legally and ethically, there is no difference between withholding a device and withdrawing a device. Legally, carrying out a request to withdraw life-sustaining treatment is neither physician-assisted suicide nor euthanasia.
DISCUSSION SHOULD BEGIN EARLY AND SHOULD BE ONGOING
The discussion of ICD deactivation should begin before the device is implanted and should continue as the patient’s health status changes. In a survey, 40% of patients said they felt that ICD deactivation should be discussed before the device is implanted, and only 5% felt that this discussion should be undertaken in the last days of life.8
At the least, it is important to identify patients with ICDs on admission to hospice and to have policies in place that ensure adequate patient education to make an informed decision about ICD deactivation at the end of life.
The topic should be discussed when goals of care change and when do-not-resuscitate status is addressed, and also when advanced directives are being acknowledged. If the patient or his or her legal representative wishes to keep the ICD turned on, that wish should be respected. The essence of a discussion is not to impose the providers’ choice on the patient, but to help the patient make the right decision for himself or herself. Of note, patients entering hospice do not have to have do-not-resuscitate status.
We believe that device management in end-of-life circumstances should be part of the discussion of the goals of care. Accordingly, healthcare providers need to be familiar with device management and to have a higher comfort level in addressing such sensitive topics with patients facing the end of life, as well as with their families.
It is also advisable to apply protocols within hospice services to address ICD management options for the patient and the legal representative. An early decision regarding end-of-life deactivation will help patients avoid distressing ICD discharges and the related emotional distress in their last moments.
- Barsheshet A, Moss AJ, Huang DT, McNitt S, Zareba W, Goldenberg I. Applicability of a risk score for prediction of the long-term (8-year) benefit of the implantable cardioverter-defibrillator. J Am Coll Cardiol 2012; 59:2075–2079.
- Kapa S, Mueller PS, Hayes DL, Asirvatham SJ. Perspectives on withdrawing pacemaker and implantable cardioverter-defibrillator therapies at end of life: results of a survey of medical and legal professionals and patients. Mayo Clin Proc 2010; 85:981–990.
- Goldstein N, Carlson M, Livote E, Kutner JS. Brief communication: management of implantable cardioverter-defibrillators in hospice: a nationwide survey. Ann Intern Med 2010; 152:296–299.
- Marinskis G, van Erven L; EHRA Scientific Initiatives Committtee. Deactivation of implanted cardioverter-defibrillators at the end of life: results of the EHRA survey. Europace 2010; 12:1176–1177.
- Kinch Westerdahl A, Sjoblom J, Mattiasson AC, Rosenqvist M, Frykman V. Implantable cardioverter-defibrillator therapy before death: high risk for painful shocks at end of life. Circulation 2014; 129:422–429.
- Goldstein NE, Lampert R, Bradley E, Lynn J, Krumholz HM. Management of implantable cardioverter defibrillators in end-of-life care. Ann Intern Med 2004; 141:835–838.
- Lampert R, Hayes DL, Annas GJ, et al; American College of Cardiology; American Geriatrics Society; American Academy of Hospice and Palliative Medicine; American Heart Association; European Heart Rhythm Association; Hospice and Palliative Nurses Association. HRS expert consensus statement on the management of cardiovascular implantable electronic devices (CIEDs) in patients nearing end of life or requesting withdrawal of therapy. Heart Rhythm 2010; 7:1008–1026.
- Raphael CE, Koa-Wing M, Stain N, Wright I, Francis DP, Kanagaratnam P. Implantable cardioverter-defibrillator recipient attitudes towards device activation: how much do patients want to know? Pacing Clin Electrophysiol 2011; 34:1628–1633.
- Barsheshet A, Moss AJ, Huang DT, McNitt S, Zareba W, Goldenberg I. Applicability of a risk score for prediction of the long-term (8-year) benefit of the implantable cardioverter-defibrillator. J Am Coll Cardiol 2012; 59:2075–2079.
- Kapa S, Mueller PS, Hayes DL, Asirvatham SJ. Perspectives on withdrawing pacemaker and implantable cardioverter-defibrillator therapies at end of life: results of a survey of medical and legal professionals and patients. Mayo Clin Proc 2010; 85:981–990.
- Goldstein N, Carlson M, Livote E, Kutner JS. Brief communication: management of implantable cardioverter-defibrillators in hospice: a nationwide survey. Ann Intern Med 2010; 152:296–299.
- Marinskis G, van Erven L; EHRA Scientific Initiatives Committtee. Deactivation of implanted cardioverter-defibrillators at the end of life: results of the EHRA survey. Europace 2010; 12:1176–1177.
- Kinch Westerdahl A, Sjoblom J, Mattiasson AC, Rosenqvist M, Frykman V. Implantable cardioverter-defibrillator therapy before death: high risk for painful shocks at end of life. Circulation 2014; 129:422–429.
- Goldstein NE, Lampert R, Bradley E, Lynn J, Krumholz HM. Management of implantable cardioverter defibrillators in end-of-life care. Ann Intern Med 2004; 141:835–838.
- Lampert R, Hayes DL, Annas GJ, et al; American College of Cardiology; American Geriatrics Society; American Academy of Hospice and Palliative Medicine; American Heart Association; European Heart Rhythm Association; Hospice and Palliative Nurses Association. HRS expert consensus statement on the management of cardiovascular implantable electronic devices (CIEDs) in patients nearing end of life or requesting withdrawal of therapy. Heart Rhythm 2010; 7:1008–1026.
- Raphael CE, Koa-Wing M, Stain N, Wright I, Francis DP, Kanagaratnam P. Implantable cardioverter-defibrillator recipient attitudes towards device activation: how much do patients want to know? Pacing Clin Electrophysiol 2011; 34:1628–1633.

Does stenting of severe renal artery stenosis improve outomes compared with medical therapy alone?
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
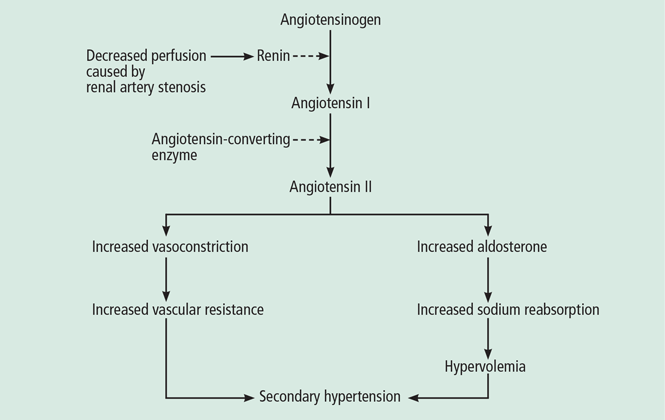
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
When does pericarditis merit a workup for autoimmune or inflammatory disease?
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
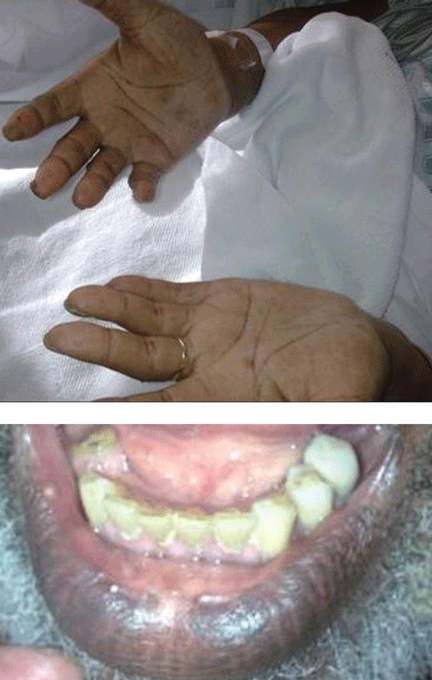
Hyperpigmentation and hypotension
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
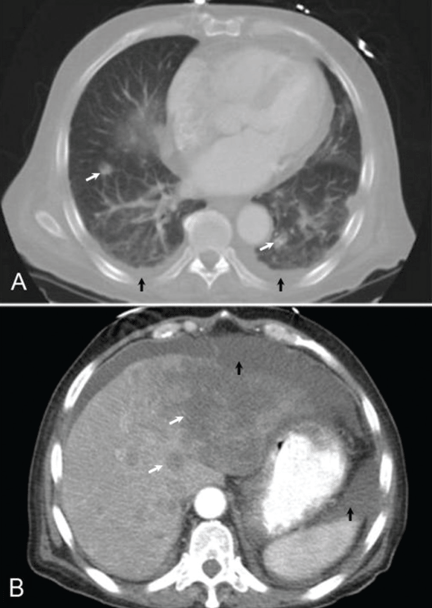
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
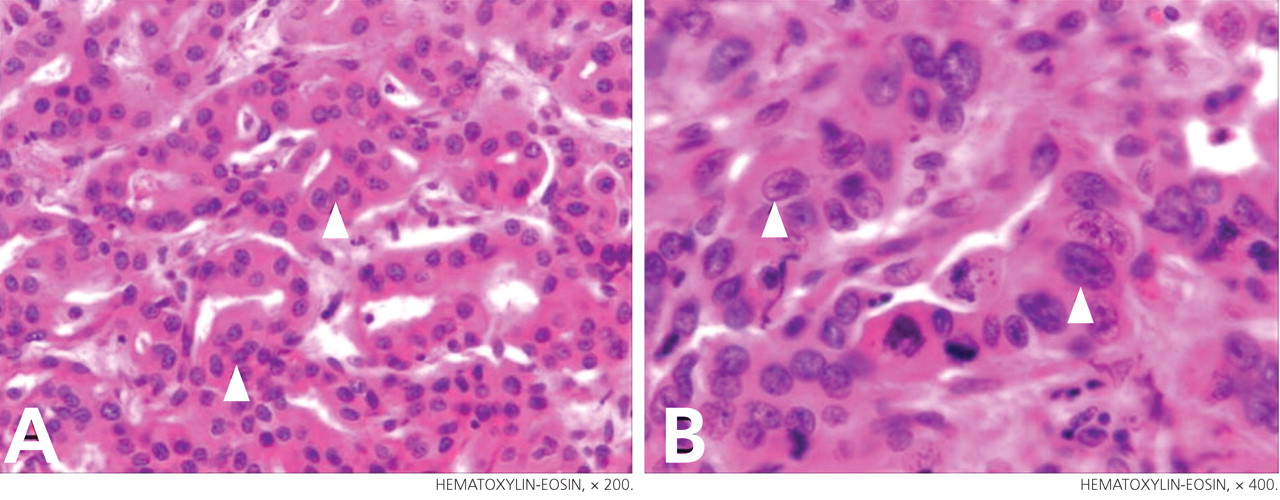
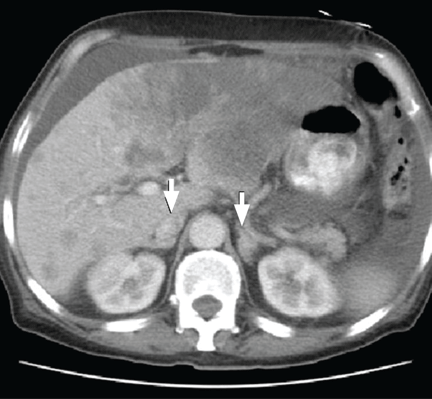
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
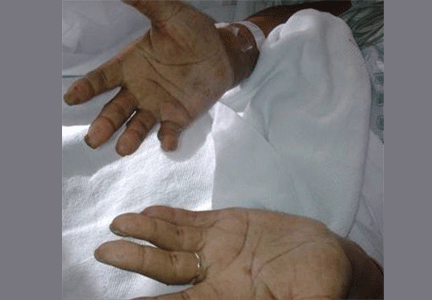
Should target natriuretic peptide levels be used for outpatient management of chronic heart failure?
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.