User login
Cutaneous and Subcutaneous Perineuriomas in 2 Pediatric Patients
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
. Reference bar indicates 500 µm.")
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
(original magnification ×15,000).")
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
. Reference bar indicates 100 µm.")
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
. Reference bar indicates 100 µm.")
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
Practice Points
- Perineuriomas are rare benign peripheral nerve sheath tumors that most commonly occur in young to middle-aged adults but rarely can present in children.
- Immunohistochemically, perineuriomas show positive staining with epithelial membrane antigen, GLUT1, claudin-1, and frequently with CD34; they are negative for S-100 and glial fibrillary acidic protein.
- Perineuriomas should be considered in the differential diagnosis in children who present with a well-circumscribed nodular lesion in the subcutaneous tissue.
(original magnification ×15,000).")
Mucous Membrane Pemphigoid Involving the Trachea and Bronchi: An Extremely Rare and Life-Threatening Presentation
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.
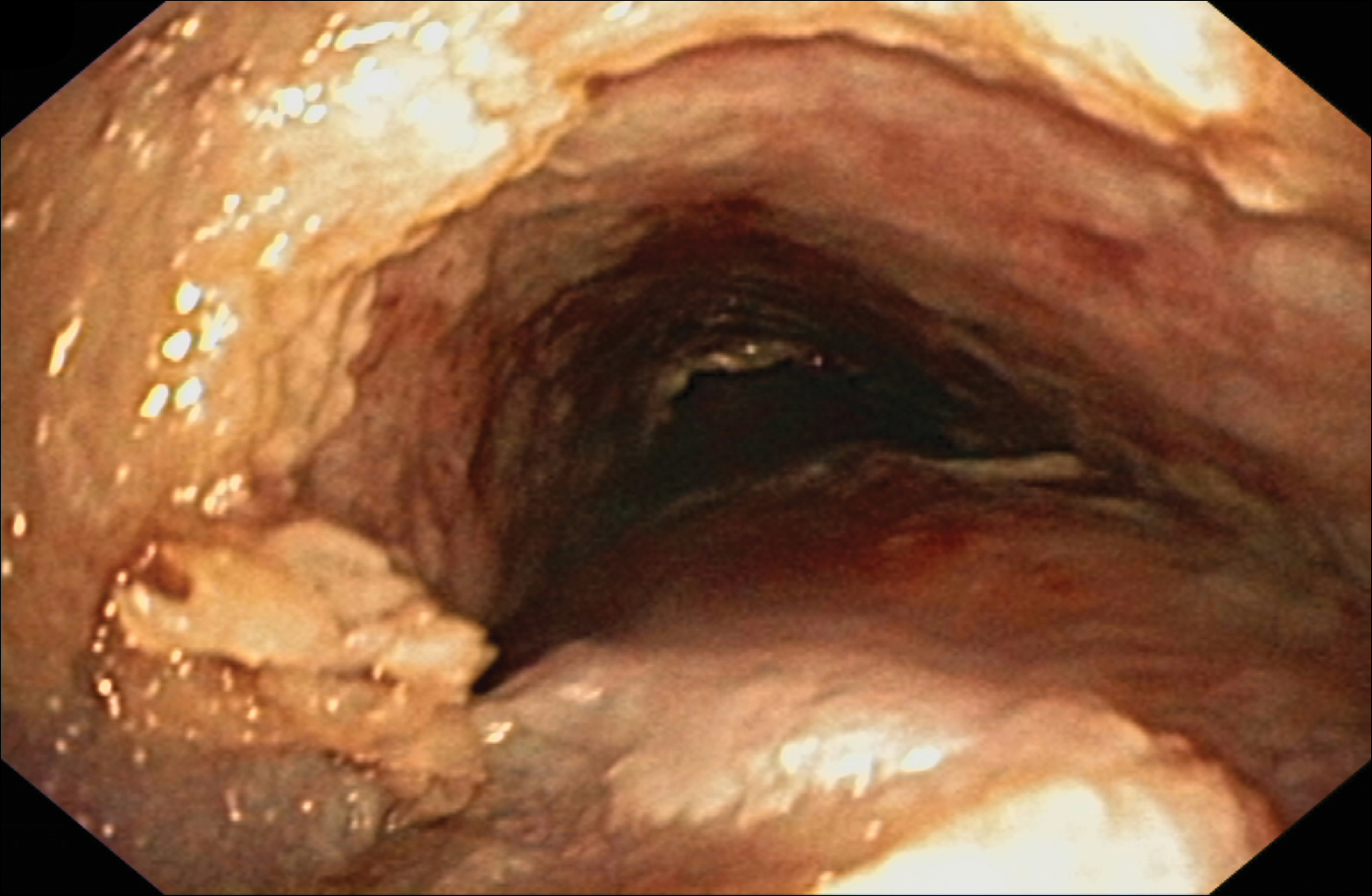

Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.
Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.
Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
Practice Points
- Mucous membrane pemphigoid (MMP) can present with diverse clinical manifestations, making the diagnosis challenging for many clinicians, including experienced dermatologists.
- If not treated early and aggressively, MMP can lead to scarring and is a potentially life-threatening disease.

