Article
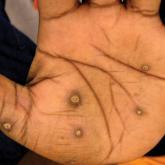
North American Blastomycosis in an Immunocompromised Patient
Blastomycosis generally produces a pulmonary form of the disease and, to a lesser extent, extrapulmonary forms, such as cutaneous, osteoarticular...

Article
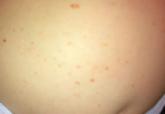
Congenital Self-healing Reticulohistiocytosis: An Underreported Entity
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a group of rare disorders characterized by the continuous replication of a...