Article
Langerhans Cell Histiocytosis Arising From a BCC: A Case Report and Review of the Literature
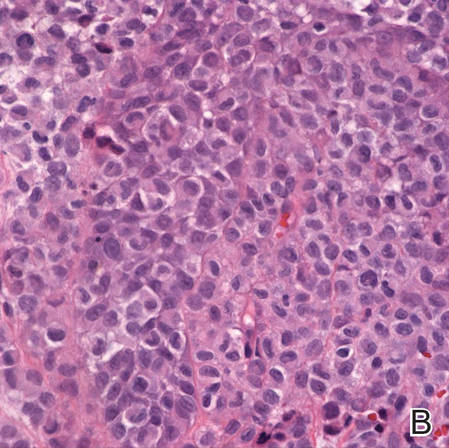
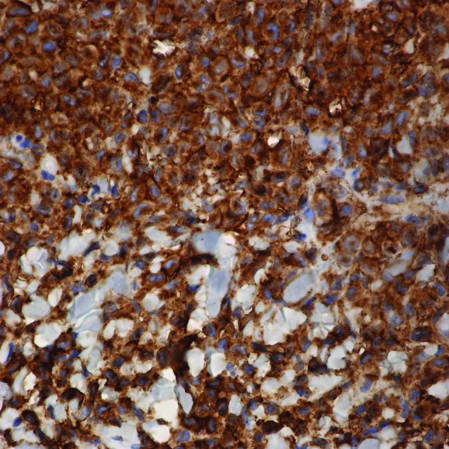
Langerhans cell histiocytosis (LCH) is a rare disease characterized by a proliferation of Langerhans cells. Several organs may be involved,...
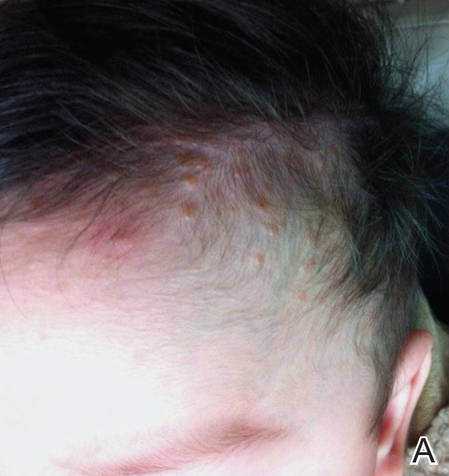
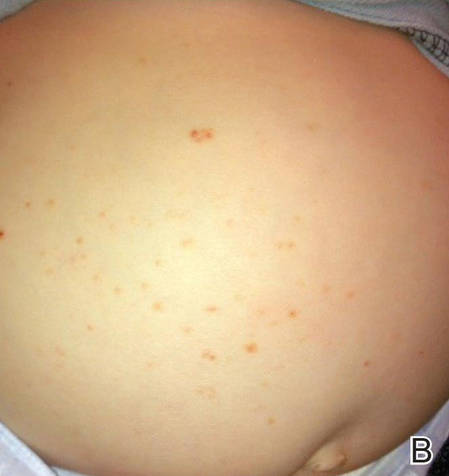
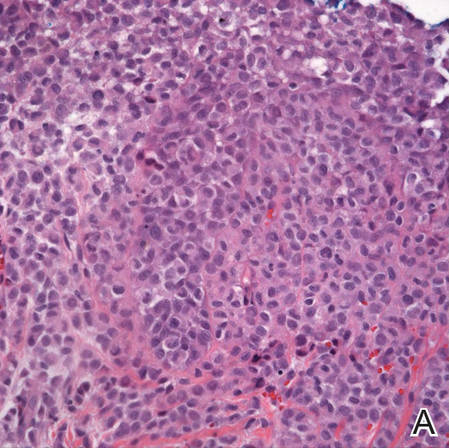
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a group of rare disorders characterized by the continuous replication of a particular white blood cell called Langerhans cells. These cells are derived from the bone marrow and are found in the epidermis, playing a large role in immune surveillance and the elimination of foreign substances from the body. Additionally, Langerhans cells are capable of migrating from the skin to lymph nodes, and in LCH, these cells begin to congregate on the bone, particularly in the head and neck region, causing a multitude of problems. Langerhans cell histiocytosis is classified into 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma. Despite various clinical presentations and severity, all subtypes are pathologically caused by the proliferation of the Langerhans cell.
Practice Points
Langerhans cell histiocytosis (LCH) is a rare disease characterized by a proliferation of Langerhans cells. Several organs may be involved,...

Adult-type Langerhans cell histiocytosis (ALCH) is characterized as a group of disorders associated with abnormal spread and proliferation of...

Multicentric reticulohistiocytosis (MR) is a rare debilitating disease that involves the skin and joints. It most commonly affects white...